这次要练习的文章数据出自:
Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 2018 Dec 4;9(1):5150.
(一)原始数据下载
首先,在GEO网站搜索:GSE111229,可以看到这里有768个原始数据。
看到图里的SRA数据的SRP133642,copy下来,然后去https://www.ebi.ac.uk/这里网站,这里可以下载SRA文件和fastq文件。

把刚才查到的SRP133642输入进去,点击搜索。
得到以下的页面:

点击SRP133642(source前面的那个小号的,不是上面的大号字)
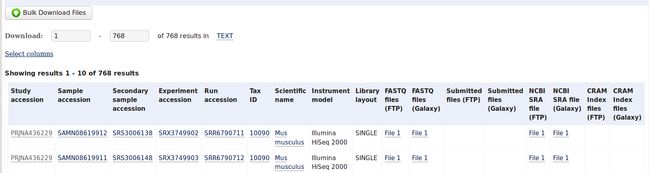
你就可以看到700多个原始文件的列表,分了好多页。这时可以点击Select columns选择你想要下载的一些选项,你可以只下载它们的编号,也可以选择下载它们的下载地址的链接,还有你可以选择下载SRA文件还是fastq文件。这里我只需要它们的fastq下载地址的链接,选好之后点击768 results后面的TEXT,然后下载下来的是一个txt文件,打开后是这样的:
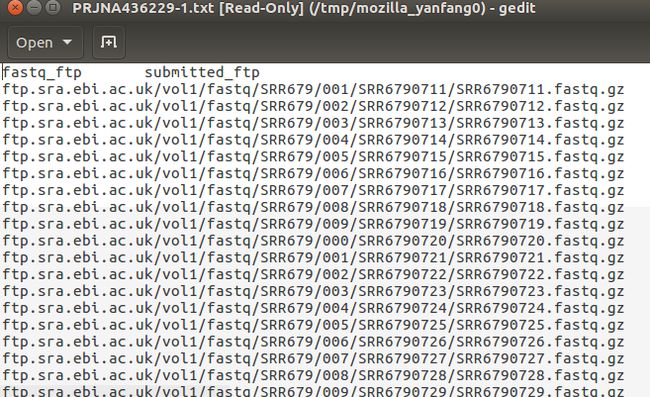
这是所有文件的fastq文件类型下载的链接地址,因为我之后要用ascp下载。
输入代码,可以先看一下前几行:
$ head fastqid.txt
ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/001/SRR6790711/SRR6790711.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/002/SRR6790712/SRR6790712.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/003/SRR6790713/SRR6790713.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/004/SRR6790714/SRR6790714.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/005/SRR6790715/SRR6790715.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/006/SRR6790716/SRR6790716.fastq.gz
接下来把每一行的开头部分“ftp.sra.ebi.ac.uk”去掉,这里就需要用到sed。把每一行的开头部分都替换成空的字符,并且替换好的输入到一个新的文件里:
$ sed -e 's/ftp.sra.ebi.ac.uk//g' fastqid.txt >> fastqid_trim.txt
再查看一下新的文件的前几行,是不是开头部分都去掉了:
$ head fastqid_trim.txt
/vol1/fastq/SRR679/001/SRR6790711/SRR6790711.fastq.gz
/vol1/fastq/SRR679/002/SRR6790712/SRR6790712.fastq.gz
/vol1/fastq/SRR679/003/SRR6790713/SRR6790713.fastq.gz
/vol1/fastq/SRR679/004/SRR6790714/SRR6790714.fastq.gz
/vol1/fastq/SRR679/005/SRR6790715/SRR6790715.fastq.gz
然后就是用ascp批量下载文件了:
$ ascp -QT -l 300m -P33001 -k1 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh --mode recv --host fasp.sra.ebi.ac.uk --user era-fasp --file-list fastqid_trim.txt /media/yanfang/FYWD/RNA_seq/fastq_files/single_cell_data/
一些参数的解释:
-T: 取消加密,否则有时候数据下载不了。
-Q:一般都加,我也不知道啥意思。
-l 设置最大传输速度,一般200m到500m。
-P33001: UDP端口。
-k: 断点续传,一般设置为值1。
-i: 提供私钥文件的地址。就是你的*.openssh这个文件放在哪里了。
--mode:模式,是上传还是下载。可以选send, recv。
--host:服务器
--user:用户
--file-list:你要下载的链接地址存在了哪个文件里,这个文件的名字。
最后要写明你下载的这些文件想存放在哪个文件夹里。
NOTE:这里提一句,如果运行ascp的时候出现了Session Stop的报错,可以尝试以下解决方法(参考:从NCBI-SRA和EBI-ENA数据库下载数据):
第一、把ascp 的-l 不要设置的太大了,一般设置为500m就可以了;(实际上我只用了300m,我这个破电脑。。。)
第二、在root权限下设置udp端口:
iptables -I INPUT -p udp --dport 33001 -j ACCEPT
iptables -I OUTPUT -p udp --dport 33001 -j ACCEPT
一开始我这两条都试了,但还是出错,整的我要崩溃,弄了一个多小时,就是一直报错,结果后来发现,是我最后的输出文件的途径写错了。。。文件夹的名字少了一个字母。。。好吧,我这个智商真是非常感人了。
写好命令行,就可以愉快的下载了,这篇文章是单端测序,所以每一个样品的Fastq文件只有一个:
$ ascp -QT -l 300m -P33001 -k1 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh --mode recv --host fasp.sra.ebi.ac.uk --user era-fasp --file-list fastqid_trim.txt /media/yanfang/FYWD/RNA_seq/fastq_files/single_cell_data/
SRR6790711.fastq.gz 100% 12MB 19.9Mb/s 00:06
SRR6790712.fastq.gz 100% 12MB 15.4Mb/s 00:13
SRR6790713.fastq.gz 100% 6453KB 14.1Mb/s 00:18
SRR6790714.fastq.gz 100% 6633KB 18.3Mb/s 00:21
SRR6790715.fastq.gz 100% 8949KB 15.7Mb/s 00:26
SRR6790716.fastq.gz 100% 6274KB 17.1Mb/s 00:31
SRR6790717.fastq.gz 100% 13MB 19.7Mb/s 00:38
SRR6790718.fastq.gz 100% 5129KB 22.5Mb/s 00:41
SRR6790719.fastq.gz 100% 10MB 25.4Mb/s 00:46
...中间的部分我就不贴上来了,700多行呢,最后下载好会有一个显示:
Completed: 8002176K bytes transferred in 4628 seconds
(14163K bits/sec), in 768 files.
(二)质控
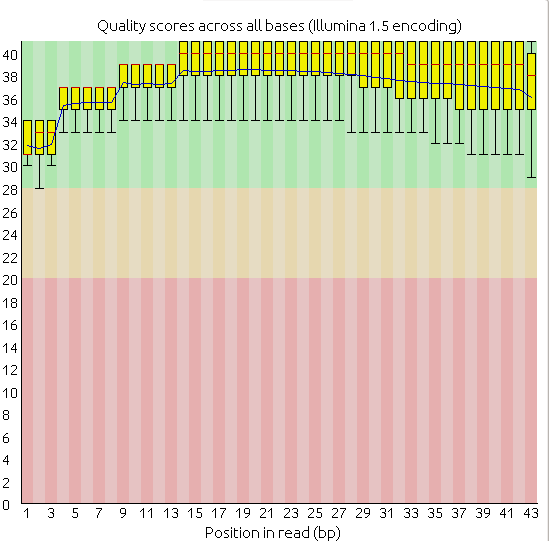
理论上要看每个样品的质量如何,决定是否要进行trim,因为这篇文章是跟着“单细胞天地”微信公众号跑一遍,主要是先大体的了解一下单细胞测序分析的过程,所以这次就没有做trim,而且我挑了一些样品做fastqc,发现总体质量还凑合,基本上都是这样的,而且没有接头污染,接头的含量显示为0。
(三)比对
在单细胞天地的公众号文章里(单细胞转录组上游分析之shell回顾 ),作者用的是hisat2进行比对的。而这篇文献的作者在方法的部分对测序分析的过程是这样描述的:“Single-end 43 bp long reads were aligned at ESCG to mm10 mouse genome using STAR v2.3.043 with default settings”。看来作者用的是STAR来进行比对的。那么我就顺便又查了一下hisat2和STAR有什么区别。根据大神的说法(https://www.jianshu.com/p/479c7b576e6f):
HISAT2找到junction正确率最高,但是在总数上却比TopHat和STAR少。HISAT2的二类错误(纳伪)比较少,但是一类错误(弃真)就高起来。就唯一比对而言,STAR是三者最佳的,主要是因为它不会像TopHat和HISAT2一样在PE比对不上的情况还强行把SE也比对到基因组上。而且在处理较长的read和较短read的不同情况,STAR的稳定性也是最佳的。就速度而言,HISAT2比STAR和TopHat2平均快上2.5~100倍。
嗯。。。看到最后一句,我还是决定用hisat2吧。。。我这破电脑如果用STAR去比对700多个样品的话,估计会冒烟。
另外,这篇文献的单细胞测序是使用了spike-in作为control的。目的是用来消除一些因素对差异分析的影响。文献中提到:“sequenced using the Smart-seq2 protocol with exogenous RNA controls from External RNA Controls Consortium (ERCC) spiked into the cell lysates”,可以看到作者是使用的spike-in RNA的。如果存在spike-inRNA,这些序列就要在比对前(构建基因组index)的时候加上去。在之后的read count步骤里,还需要把spike-in和参考基因组GTF文件合并在一起。因为外源加入的spike-in的物种不是小鼠的,如果不在比对的时候加入到参考基因组里,那么你比对出来所有的spike-in是不会有任何Read比对到参考基因组上的,那么spike-in的意义就没有了。我下载的是mm10基因组的fasta文件和GTF文件,ERCC(spike-in)的下载地址是:https://www.thermofisher.com/search/results?query=ERCC92.gtf%20&focusarea=Search%20All。合并的代码(https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms_095048.txt):
$ cat ERCC92.fa >> mm10_plus_spikein.fa
$ cat ERCC92.gtf >> gencode.vM22.annotation_plus_ERCC.gtf
合并之后需要自己构建参考基因组索引,因为我之前下载的小鼠的索引是没有spike-in的,现在加上了spike-in,所以要自己构建了。代码很简单:
$ hisat2-build -p 20 mm10_plus_spikein.fa genome
构建索引的速度取决于你的电脑,我这个破电脑用了2个多小时。也还算可以接受的。
下面就开始比对了:
ls *.gz | while read i;do hisat2 -p 10 -x /media/yanfang/FYWD/RNA_seq/ref_genome/index/mm10_plus_ERCC_hisat2/genome -U $i -S /media/yanfang/FYWD/RNA_seq/sam_files/single_cell_sam/${i%%.*}.sam;done
402682 reads; of these:
402682 (100.00%) were unpaired; of these:
55003 (13.66%) aligned 0 times
347439 (86.28%) aligned exactly 1 time
240 (0.06%) aligned >1 times
86.34% overall alignment rate
395135 reads; of these:
395135 (100.00%) were unpaired; of these:
50638 (12.82%) aligned 0 times
344300 (87.13%) aligned exactly 1 time
197 (0.05%) aligned >1 times
87.18% overall alignment rate
212554 reads; of these:
212554 (100.00%) were unpaired; of these:
55914 (26.31%) aligned 0 times
156545 (73.65%) aligned exactly 1 time
95 (0.04%) aligned >1 times
73.69% overall alignment rate
......
参数解释:
这里用了一个while语句,关于while语句,可以参考我之前贴上来的文章(结构化命令 之 while命令)。
-P 10:用10个线程。
-x:后面跟的是小鼠基因组index的存放位置。
-U:单端数据文件。
-S:输出文件存放的位置。
%%.*:取输入原始测序文件(i)的前缀。
(四)sam转换成bam文件
$ ls *.sam | while read i ;do (samtools sort -O bam -@ 10 -o /media/yanfang/FYWD/RNA_seq/bam_files/single_cell_bam/$(basename ${i} ".sam").bam ${i});done
其中一个小tip就是:basename这里,会返回每个$i,也就是每个sam文件的名称,然后".sam"就是将名称中的.sam去掉,于是只留下了前缀名,这样才能更改文件名为bam。(这个方法来自:单细胞转录组上游分析之shell回顾 )
(五)构建索引
接下来就是构建每一个bam文件的索引了~
$ ls *.bam |xargs -i samtools index {}
这里我查了一下xargs是什么命令:
xargs 可以将管道或标准输入(stdin)数据转换成命令行参数,也能够从文件的输出中读取数据。xargs 是一个强有力的命令,它能够捕获一个命令的输出,然后传递给另外一个命令。之所以能用到这个命令,关键是由于很多命令不支持|管道来传递参数,所以就有了 xargs 命令。一般是和管道一起使用。
命令格式:
somecommand |xargs -item command
(六)count计数
之前我自己练习的bulk-RNA-seq分析时,这一步read计数我都是使用的htseq-count这个软件。而这次看单细胞天地文章的时候,作者在进行read count时使用了featureCounts。原谅我的无知。。。之前没有听说过这个软件,于是上网搜索了featureCounts和htseq-count的区别:
(1)首先,这两个软件运行后read count数是不一样的。(例如:htseqcount和featurecount计数不一样 )
(2) HTSeq-count对于多重比对的reads则是采取舍弃策略。(参考转录组定量可以用替换featureCounts代替HTSeq-count)
(3) Htseq-count慢!如果样本数是几百个时,其所消耗的时间是以天记的。(怪不得我之前6个8个的样本就要过夜跑。。。也不排除我这破电脑的原因)
(4)不仅支持基因或转录本的定量,还可以进行exons, promoter, gene bodies, genomic bins and chromosomal locations的定量。
“单细胞天地”给出的代码是:
# 还是先指定gtf的位置
$ gtf=/YOUR_GTF_PATH/
$ featureCounts -T 5 -t exon -g gene_id -a $gtf -o all.id.txt *.bam 1>counts.id.log 2>&1 &
# 如果是双端测序,可以加上-p选项
-T:使用的线程数。
-t:Specify feature type in GTF annotation. `exon' by default. Features used for read counting will be extracted from annotation using the provided value.要计数的feature名称,也就是GTF的第三列信息,默认是exon。
-a:Name of an annotation file.指定使用的注释文件。
-o: Name of the output file including read counts. 输出文件。
-g :Specify attribute type in GTF annotation. "gene_id" by default. Meta-features used for read counting will be extracted from annotation using the provided value.提取的GTF最后一列attribute信息,默认是gene_id。如果你不想让你的count 结果是ENSMUSG。。。一堆数字,而是基因的具体名称,则需要把gene_id换成gene_name。
下面根据我的文件位置,更改一下代码:
$ featureCounts -T 5 -t exon -g gene_id -a /media/yanfang/FYWD/RNA_seq/ref_genome/gencode.vM22.annotation_plus_ERCC.gtf -o /media/yanfang/FYWD/RNA_seq/matrix/singlecell/all.id.txt *.bam 1>counts.id.log 2>&1
这里说一句,1>counts.id.log的意思是标准输出;2>&1 的意思就是将标准错误重定向到标准输出。可以把counts.id.log看作"黑洞",它等价于一个只写文件,所有写入它的内容都会永远丢失,而尝试从它那儿读取内容则什么也读不到。
最后得到两个文件:一个是矩阵,另外一个是矩阵的总结信息
(七)过滤表达矩阵
得到矩阵以后,先读取矩阵
> a=read.table('all.id.txt',header = T ,sep = '\t')
单细胞测序嘛,肯定很多基因的表达量都是0,比如说矩阵的第一行:
> x=a[1,]#把第一行赋值给x
> table(x>1)#看一眼第一行里这个基因表达量大于1的细胞有多少个
FALSE TRUE
768 5
#结果在这么多的细胞里,只有5个细胞有这个基因的表达
那我们有700多个细胞,总不能一个一个的看吧?所以要用到循环。循环的具体代码完全按照这篇文章的来:获取Github代码包以及准备工作。这篇文章的代码也是解析的非常详细。
>floor(ncol(a)/50)#用总列数除以50然后向下取整,结果就是15
也就是说,只要一行中至少要在15个样本中有表达量。这个对矩阵的过滤,没有统一的标准。我查询了几篇文章,几篇文章里的过滤标准都不一样。有的是按照每一个细胞里的基因数过滤的(有的人要求一个细胞里要有7000个基因表达,有的人100就够了),有的是按照每一个基因在一定的样品数里有表达,这个就不一定了,要看你的样品量和你的分析目的。我特意贴了三篇对矩阵过滤的文章,可以看出这一步完全看心情???比如下面这篇:
使用limma、Glimma和edgeR,RNA-seq数据分析易如反掌
“在任何样本中都没有足够多的序列片段的基因应该从下游分析中过滤掉。这样做的原因有好几个。 从生物学的角度来看,在任何条件下的表达水平都不具有生物学意义的基因都不值得关注,因此最好忽略。 从统计学的角度来看,去除低表达计数基因使数据中的均值 - 方差关系可以得到更精确的估计,并且还减少了在观察差异表达的下游分析中需要进行的统计检验的数量。”
还有这篇:
单细胞RNA-seq分析
1.去除在任何细胞中都不表达的基因。
2.具有少量reads/molecules,很可能是已经被破坏或捕获细胞失败,应该移除这类细胞
3.手动过滤细胞:大多数细胞检测到的基因在7000-10000之间,这对于高深度scRNAseq来说是正常的。但是这也取决于实验协议和测序深度。比如说基于droplet或其他测序深度较低的方法,通常其每个细胞检测到的基因较少。如果细胞检出率相等,则分布应近似正常,因此我们移除分布尾部的那些细胞(检测到的基因少于7000的细胞)
4.自动过滤细胞:自动异常检测来识别可能存在问题的细胞:scater包创建一个矩阵(行代表cell,列代表不同的QC度量),然后PAC提供了按照QC度量排序的cells的2D表示,然后使用来自mvoutlier包的方法检测异常值。在这里插入图片描述
5.过滤基因:移除表达水平被认为是undetectable的基因。We define a gene as detectable if at least two cells contain more than 1 transcript from the gene. If we were considering read counts rather than UMI counts a reasonable threshold is to require at least five reads in at least two cells. 然而很多时候阈值取决于测序深度,而且很重要的一点是基因过滤一定要在细胞过滤之后,因为一些基因可能仅仅被检测到只存在在低质量的细胞里。
还有一篇是这样写的:
单细胞测序(scRNA-seq)通关||数据处理必知必会
根据基因的表达量等特征,对细胞进行过滤,通常的做法就是指定一个阈值,比如要求一个细胞中检测到的基因数必须大于100,才可以进入到下游分析,如果小于这个数字,就过滤掉该细胞。需要强调的是,在设定过滤的阈值时,需要人为判断,这样的设定方式会受到主观因素的干扰,所以往往都会指定一个非常小的过滤范围,保证只过滤掉极少数的离群值点。
综上所述,矩阵过滤没有统一标准,请自行尝试。
继续说矩阵的过滤代码:
上面知道了 x>1 返回逻辑值0和1,0为FALSE,1为TRUE。现在我们要找一行中总共有多少TRUE,就用sum计算一下(因为会忽略掉0的影响)
>sum(x>1) > floor(ncol(a)/50)# 当然第一行会返回FALSE,也就表明我们要去掉这一行内容
>a[sum(x>1) > floor(ncol(a)/50),]# 就把不符合要求的第一行去掉了
这是对一行的操作,那么对于上万行的矩阵怎么操作?答:用apply()
> allsample=a[apply(a,1, function(x) sum(x>1) > floor(ncol(a)/50)),]
#首先,对a这个矩阵进行操作
#其次,对行(也就是1表示)进行操作;如果对列操作,用2表示。
#复杂的操作先写上 function(x){},这是一个标准格式。
#最后,把筛选完成的矩阵赋值给新的变量。
> dim(allsample)#看看新的矩阵里保留了多少基因?16289个。
[1] 16289 773
(八)过滤后操作
过滤之后,看一下样品里的spike-in的情况(有关spike-in的介绍,这篇文章里的几篇文献有提到单细胞RNA-seq入门文献学习):
> grep('ERCC',rownames(allsample))
integer(0)
简直惊呆了我!明明有92个spike-in的control,怎么一个也没有?后来我看了一下表达矩阵才发现:基因名,或者说spike-in的名字,被当成了第一列,并没有成为行名,所以才检索不到有spike-in的存在。那么需要把第一列设置成行名:
> rownames(a)<-a[,1] #将数据框的第一列作为行名
> b<-a[,-1] #将数据框的第一列删除,只留下剩余的列作为数据
然后就可以看spike-in的情况了:
> grep('ERCC',rownames(allsample))
[1] 15928 15929 15930 15931 15932 15933 15934 15935 15936 15937 15938 15939 15940 15941 15942 15943 15944 15945 15946 15947
[21] 15948 15949 15950 15951 15952 15953 15954 15955 15956 15957 15958 15959 15960 15961 15962 15963 15964 15965 15966 15967
[41] 15968 15969 15970 15971 15972 15973 15974 15975 15976 15977 15978 15979 15980 15981 15982 15983 15984 15985 15986 15987
[61] 15988 15989
(九)去除文库大小差异(参考:常说的表达矩阵,那得到之后呢?)
每个细胞测得数据大小不同,这样是没办法看高表达还是低表达的,必须先保证基数一样才能比较,cpm(counts per million)这个算法就是做这个事情的。
cpm是归一化的一种方法,代表每百万碱基中每个转录本的count值。
注意:这个算法只是校正文库差异,而没有校正基因长度差异。要注意我们分析的目的就是:比较一个基因在不同细胞的表达量差异,而不是考虑一个样本中不同两个基因的差异,因为"没有两片相同的树叶”这个差异是正常的。但是同一个基因由于某种条件发生了改变,背后的生物学意义是更值得探索的。
用起来很简单,有现成的函数cpm() ,然后我们再用log将数据降个维度,但保持原有数据形状不变:log2(edgeR::cpm(dat)+1) 。
意思就是:cpm需要除以测序总reads数,而这个值作为分母会导致结果千差万别,有的特别大有的很小。为了后面可视化不受极值的影响,用log转换一下可以将数值变小,并且原来大的数值最后还是大,并不改变这个现实。
# 先看看前6行6列的数据
>finaldata[1:6,1:6]
X.SS2_15_0048_A1 X.SS2_15_0048_A2 X.SS2_15_0048_A3 X.SS2_15_0048_A4 X.SS2_15_0048_A5 X.SS2_15_0048_A6
ENSMUSG00000102269.1 0 0 0 0 0 0
ENSMUSG00000098104.1 5 0 0 0 1 1
ENSMUSG00000103922.1 1 0 1 0 0 0
ENSMUSG00000033845.13 0 0 9 1 19 0
ENSMUSG00000025903.14 202 46 29 80 0 27
ENSMUSG00000033813.15 1 5 1 0 9 8
# 比如先计算一下第五个样本的总测序量
> sum(dat[,5])
[1] 215937 #说明第五个样品有这么多的read数
# 那么对于第5个样品的第4个基因read数是19
# 计算它的cpm值:count值*1000000/总测序reads
> 19*1000000/215937
[1] 87.98863
# 和标准公式比较看看,结果完全相同
> edgeR::cpm(finaldata[,5])[4]
[1] 87.98863
# 因此最后就是
> finaldata2=log2(edgeR::cpm(finaldata)+1)
#把去除文库大小差异后的matrix保存成一个table
> write.table(finaldata2,"adjust_library_matrix.txt",sep="\t")