Bioinformatics: Assembling Genomes (week 1-2)
Bioinformatics: Assembling Genomes (week 1-2)
本文为Coursera课程 Assembling Genomes and Sequencing Antibiotics (Bioinformatics II) 笔记。
作者:ybw
- Bioinformatics Assembling Genomes week 1-2
- Introduction
- Motivation
- Problems
- Terminologies
- The ideal model - From Hamilton to de Bruijn
- The ideal model
- From a k-mers to a graph
- Using Grpahs to assemble - Hamiltonian Path
- From Hamiltonian Path to Eulerian Path
- k-universal string
- de Bruijn Graph
- Eulerian Paths
- Eulerian Cycle and Eulerian Path
- Existence
- Code
- Code Analysis
- Eulerian Cycle Based on Stack
- Eulerian Path
- Solve the String Reconstruction Problem
- k-Universal Circular String Problem
- Advanced Methods
- Assembling Genomes from Read-Pairs
- String Reconstruction from Read-Pairs Problem
- Breaking reads into k-mers
- Splitting the genome into contigs
- Assembling error-prone reads
- Introduction
Introduction
Motivation
To sequence a genome:
1. 目前缺少读取完整DNA链的技术;
2. 采用将DNA链打碎成reads并识别的方法(荧光标记法)来获取DNA信息;
3. 如何将reads拼接成原来的DNA。
具体过程如图1所示, 将包含无数个相同DNA的组织样本或血样使用生物学方法打碎成短的fragments,并识别这些fragments,从而得到reads。
图1:

Problems
- DNA为双链结构(double-stranded),无法分辨每个read属于哪个链;
- 目前识别reads的方法并不完美,其中的氨基酸和DNA链中可能并非完全相同;
- DNA中的某些区域可能没有被任何reads覆盖,导致无法完整地重构基因(genome)。
Terminologies
- read: short DNA fragments called reads。
- k-mer:
- strand: DNA单链
The ideal model - From Hamilton to de Bruijn
The ideal model
首先在理想条件研究问题。假设理想情况如下:
* 所有的reads来自同一个strand, 且没有错误;
* Perfect coverage - 任何属于该strand的k-mer都会产生reads。
From a k-mers to a graph
为了在k-mers之间建立联系,使用prefix和suffix来分别表示一个k-mer的前k-1个核苷酸和后k-1个核苷酸(nucleotide)。例如Prefix (TAA) = TA, Suffix (TAA) = AA。
为了建图,用每个k-mer来表示图中的一个node,当 prefix(kmer1)=suffix(kmer2) ,则添加一条从 kmer1 到 kmer2 的有向边。
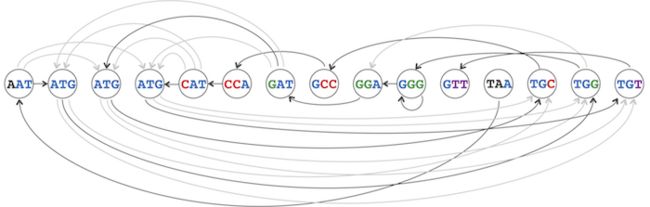
由此所建立的图称为overlap graph,记为 Overlap(Patterns) 。图2所示的是由DNA链TAATGCCATGGGATGTT所产生的k-mers组成的overlap graph,图中实线表示的路径代表基因序列“TAATGCCATGGGATGTT”。
图2-The genome path spelling out TAATGCCATGGGATGTT, highlighted in the overlap graph:
Using Grpahs to assemble - Hamiltonian Path
在overlap graph中,edges代表可能的DNA序列,例如图2中的边(TAA) -> (AAT)表示TAAT。如果可以在overlap graph中找到一个路径(path),该路径经过所有节点且只经过一次,则该路径就可以表示该图可产生的一个genome。根据定义,这条路径即是哈密顿路径。
A path in a graph visiting every node once is called a Hamiltonian path.
由于overlap graph中的Hamiltonian path不止一条,而且Hamiltonian path是NP-Complet问题,因此当问题规模太大时就无法处理了。为了解决该问题,研究人员发现了另一种方法 - de Bruijn Graph。
另外,人类基因组计划好像是通过Hamiltonian path来计算的,因为当时de Bruijn Graph并没有被发现。
From Hamiltonian Path to Eulerian Path
k-universal string
在引入de Bruijn Graph之前,我们首先看一下荷兰数学家Nicolaas de Bruijn的一项研究。1946年,de Bruijn对一个纯理论问题产生了兴趣,该问题表述如下。
A binary string is a string composed only of 0’s and 1’s; a binary string is k-universal if it contains every binary k-mer exactly once.
For example, 0001110100 is a 3-universal string, as it contains each of the eight binary 3-mers (000, 001, 011, 111, 110, 101, 010, and 100) exactly once.
当图中的节点为所有的binary k-mers时,解决k-universal string问题和解决String Reconstruction Problem问题是等价的。我们已经知道使用哈密顿路径可以解决String Reconstruction Problem,那么哈密顿路径也一定可以解决该问题。图3为一个例子。
图3:A Hamiltonian path in the overlap graph of all binary 3-mers.

然而,de Bruijn当时并没有使用哈密顿路径来解决该问题,而是用了一个不那么直观的方法来表示k-mer:
使用边而非节点来表示k-mers。
de Bruijn Graph
将该思想引入String Reconstruction Problem,就可得到de Bruijn Graph。在String Reconstruction Problem问题中,de Bruijn graph的边根据输入的k-mers来建立,而边两边的节点分别为该边所表示的k-mer的prefix和suffix。问题表述如下:
DeBruijn Graph from k-mers Problem: Construct the de Bruijn graph from a set of k-mers.
Input: A collection of k-mers Patterns.
Output: The adjacency list of the de Bruijn graph DeBruijn(Patterns).
解决步骤包括:
- 使用有向边来表示k-mer,则边的起点为该k-mer的prefix, 终点为suffix。如图4;
- 将相同节点合并,如图5;
图4:Genome TAATGCCATGGGATGTT represented as a path with edges (rather than nodes) labeled by 3-mers and nodes labeled by 2-mers.
![]()
图5:Gluing nodes

Eulerian Paths
尽管我们将de Bruijn graph中相同的nodes进行了合并,但由于图中的边并没有改变,因此图中所携带的有关genome的信息并没有发生变化。与之前类似,因为de Bruijn graph中每个边都代表一个k-mer,只需找到一个路径,经过图中所有的边且每个边只经过一次,即可解决String Reconstruction Problem问题。即,solving the String Reconstruction Problem reduces to finding an Eulerian Path in the de Bruijn graph.
注:
* 该图中的边由给出的k-mers来创建,在图建好之后,不能根据节点信息随意添加新的边;
* 图中的欧拉路径可能不止一条。
Eulerian Cycle and Eulerian Path
那么,如何从图中找到一条欧拉路径,这节将给出具体算法。
Existence
欧拉路径问题可以追述到哥尼斯堡七桥问题(Bridges of Königsberg Problem):Is it possible to set out from my house, cross each bridge exactly once, and return home? 哥尼斯堡的地图可以简化至图6。
图6:The graph Königsberg.

该问题等价于: 该图中是否存在欧拉回路(Eulerian Cycle)。
Eulerian cycle: A cycle that traverses each edge of a graph exactly once is called an Eulerian cycle.
对于无向图:
A graph has an Eulerian circuit if and only if it is connected and every node has ‘even degree’.
对有向图,为了欧拉回路是否存在,引入balance概念:
当一个节点的出度和入度相等,称该节点为balanced;
当一个图中所有的节点都为balanced,称该图为balanced。
(A node v is balanced if in(v)=out(v) , and a graph is balanced if all its nodes are balanced. )
那么是否balanced graphs一定存在欧拉回路呢? 答案是否定的,因为该图未必是强联通的(strongly connected)。
Euler’s Theorem: Every balanced, strongly connected directed graph is Eulerian.
(An Eulerian graph is a graph containing an Eulerian cycle.)
对于balanced and strongly connected directed graphs,如何找到an Eulerian cycle。下面给出算法。
Code
Pseudo-Code:
Hierholzer’s algorithm
find_cycle(node i)
if node i has no neighbors
cycle[pos] = node i
pos++
else
while(node i has neighbors)
pick a neighbor j of node i
delete_edges(node j, node i)
find_cycle(node j)
cycle[pos] = node i
pos++Python代码:
使用dict来保存adjacency list:
graph{ node1:[list_of_edges],
node2:[list_of_edges],
...}import sys
sys.setrecursionlimit(1000000)
def eulerian_cycle(graph):
start_node = 0
dfs(start_node, graph)
cycle=[]
def dfs(node, g):
global cycle
if len(g[node]) == 0:
cycle.append(node)
else:
while g[node]:
next_node = g[node].pop()
dfs(next_node, g)
cycle.append(node)
# reverse the circuit
cycle.reverse()Code Analysis
- 算法复杂度为 O(m+n) ,其中 m 为edges数量, n 为nodes数量;
- 函数eulerian_cycle最后计算的结果为实际路径的逆序;
- 该算法使用递归和全局变量cycle,对于规模较大的图,容易产生栈溢出;
- Python本身对递归最大深度有限制,使用sys.setrecursionlimit()可手动设定递归最大深度;
- 即使使用sys.setrecursionlimit,对于规模较大的图,在实际使用中,依然产生了python.exe停止运行的问题(推测原因为栈溢出)。
Eulerian Cycle Based on Stack
为了解决递归导致的栈溢出问题,使用自己的stack来实现上述算法。
def eulerian_cycle(g, start):
# use stack to emulate recursion. Avoid large number of recursions.
path = []
stack = [start]
while stack:
v = stack[-1]
if g[v]:
# if v in g has edge(s), random pop
j = random.randrange(len(g[v]))
u = g[v].pop(j)
stack.append(u)
else:
path.append( stack.pop() )
path.reverse()
return path注: path在return之前依然需要reverse。
Eulerian Path
存在条件:
对于无向图
A strongly connected graph with two odd vertices have an Euler path.
对于有向图
A nearly balanced graph has an Eulerian path if and only if adding an edge between its unbalanced nodes makes the graph balanced and strongly connected.
即当该图中只有两个nodes不平衡,而且其中一个in-degree多1,另一个out-degree多1。
因此,解决有向图的欧拉路径问题,只需检查每个node的in-degree和out-degree,找到in-degree多1的那个,并以它为起点(start_node)运行eulerian cycle算法即可。
Solve the String Reconstruction Problem
使用Eulerian path解决 String reconstruction problem:
CODE CHALLENGE: Solve the String Reconstruction Problem.
Input: An integer k followed by a list of k-mers Patterns.
Output: A string Text with k-mer composition equal to Patterns. (If multiple answers exist, you may return any one.)Answer = genomePath(EulerianPath(DeBruijnGraph(k-mers)))
k-Universal Circular String Problem
回到de Bruijn最初思考的问题上来。
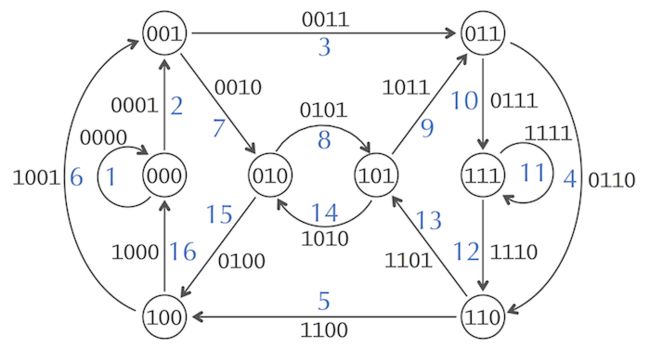
Let BinaryStringsk be the set of all 2k binary k-mers. The only thing we need to do is solve the k-Universal Circular String Problem is to find an Eulerian cycle in DeBruijn(BinaryStringsk) .
例子如图7所示。
注:
* 该问题的de Brujin graph中,nodes为所有 2k−1 个 binary (k - 1)-mers,edges为所有 2k 个k-mers;
* A directed edge connects (k - 1)-mer Pattern to (k - 1)-mer Pattern′ if there exists a k-mer whose prefix is Pattern and whose suffix is Pattern′ .
图7:An Eulerian cycle spelling the cyclic 4-universal string 0000110010111101 in DeBruijn(BinaryStrings4).
Advanced Methods
Assembling Genomes from Read-Pairs
在不存在错误的情况下,reads长度越短,de Bruijn graph越复杂,当reads足够长,de Bruijn graph很可能收敛为一条路径。如图8所示。
但技术限制,目前如法产生准确而又足够长的reads(只能精确的产生300左右个核苷酸长度的reads,对于绝大多数重复序列来说依然太短,因此实际中de Bruijn graph的欧拉路径并不唯一,为解决该问题,研究人员发明了另一种曲线救国方法——read-pairs。
Read-pair are pairs of reads separated by a fixed distance d in the genome.
Read-paris技术可以将间隔特定距离( d nucleotides)的两个k-mers联系起来,从而间接增加了reads的长度。一个典型的read-pair长度可看作 k+d+k , 记为(k,d)-mer,其中前一个 k 所表示的k-mer和后一个 k 所表示的k-mer是已知的,中间长为 d 的gap的信息是未知的(但 d 是已知的且是固定的)。
通过(3,1)-mers来重建序列 (TAATGCCATGGGATGTT) 的例子如下:
TAA GCC
AAT CCA
ATG CAT
TGC ATG
GCC TGG
CCA GGG
CAT GGA
ATG GAT
TGG ATG
GGG TGT
GGA GTT
TAATGCCATGGGATGTTString Reconstruction from Read-Pairs Problem
CODE CHALLENGE: Solve the String Reconstruction from Read-Pairs Problem.
Input: Integers k and d followed by a collection of paired k-mers PairedReads.
Output: A string Text with (k, d)-mer composition equal to PairedReads.解决该问题需要用到paired de Bruijn graph。使用 read1−read2 来表示(k,d)-mer,则图中的nodes由两部分组成,当 node1 到 node2 有一条边,代表 read1−read2 ,则有
如图8所示。
图8:A paired de Bruijn graph constructed from a collection of nine (2,1)-mers.

注意: paired de Bruijn graphs 可能存在多个欧拉路径,但并非每个欧拉路径都是合法的。
判别方法:将该路径上所有边的第一个read和第二个read分别进行重建组合,得到距离为 k+d 的两个strings,比较两个strings的重叠部分,若完全匹配,则该路径合法;否则,该路径不合法。 遍历不同的欧拉路径,直到找到合法路径为止。
例如,图8中,存在合法路径:
AG-AG
GC-GC
CA-CT
AG-TG
GC-GC
CT-CT
TG-TG
GC-GC
CT-CA
AGCAGCTGCTGCA
PrefixString = AGCAGCTGCT
SuffixString = AGCTGCTGCA
Genome = AGCAGCTGCTGCA 和非法路径
AG-AG
GC-GC
CT-CT
TG-TG
GC-GC
CA-CT
AG-TG
GC-GC
CT-CA
AGC?GC?GCTGCA
PrefixString = AGCTGCAGCT
SuffixString = AGCTGCTGCABreaking reads into k-mers
在实际条件中,无法生成genome中的所有k-mers,导致覆盖不完全(imperfect coverage)问题。
将reads划分为更短的k-mers有助于产生完美的覆盖(perfect coverage)。如下图所示。
图9:Breaking 10-mer reads (left) into 5-mers results in perfect coverage of a genome by 5-mers (right).

Trade-off:
* k 的值越小,覆盖可能越完美;
* k 的减小,会导致de Bruijn graph变得更复杂,使得通过graph来推断genome变得更困难。
Splitting the genome into contigs
在实际基因测序中,即使经过了reads breaking,也依然存在genome的某些区域没有一个reads覆盖的情况,从而导致de Bruijn graph丢失一些edges,无法产生完整的欧拉路径。此时,生物学家退而求其次,将目标定为assembling contigs 而非assembling the entire chromosomes。幸运的是,我们可以从de Bruijn graph中推导出contigs。
Definitions:
Contigs: long, contiguous segments of the genome.
A path in a graph is called non-branching if in(v) = out(v) = 1 for each intermediate node v of this path, i.e., for each node except possibly the starting and ending node of a path. 即,除了第一个和最后一个节点,路径中的其他节点的出度和入度均为1.
A maximal non-branching path is a non-branching path that cannot be extended into a longer non-branching path. (并不是图中最长的那条,而是有很多条长度各不相同的)
如下图所示。
图10:Breaking the de Bruijn graph into nine maximal non-branching paths representing contigs TAAT, TGTT,TGCCAT, ATG, ATG, ATG, TGG, GGG, and GGAT.

Pseudo-code for the max non-branching path problem.
The MaximalNonBranchingPaths pseudocode below generates all non-branching paths in a graph. It iterates through all nodes of the graph that are not 1-in-1-out nodes and generates all non-branching paths starting at each such node. In a final step, MaximalNonBranchingPaths finds all isolated cycles in the graph.
MaximalNonBranchingPaths(Graph)
Paths ← empty list
for each node v in Graph
if v is not a 1-in-1-out node
if out(v) > 0
for each outgoing edge (v, w) from v
NonBranchingPath ← the path consisting of the single edge (v, w)
while w is a 1-in-1-out node
extend NonBranchingPath by the outgoing edge (w, u) from w
w ← u
add NonBranchingPath to the set Paths
for each isolated cycle Cycle in Graph
add Cycle to Paths
return Paths该算法分为两部分:
第一个(种情况)for循环解决起点为非1-in-1-out node的情况(该情况下终点也一定是非1-in-1-out node);
第二个(种情况)for循环解决(并没有解决)一条环路循环的情况,该情况下路径中的所有nodes都是1-in-1-out node。
在实际使用中,须记录每个1-in-1-out node的访问情况。出现在第一种路径中的1-in-1-out node一定不属于第二种情况。
Python code.
def maximalNonBranchingPaths(g):
"""
Assume the type of nodes is Integer
with the value from 0 to len(g)-1.
"""
# Count the in-degree of nodes.
indegree = [0] * (len(g))
for node in g:
for v in g[node]:
indegree[v] += 1
def oneone(v):
"""
Check if the node v is a 1-in-1-out node.
True for yes, False for no.
"""
nonlocal indegree, g
if indegree[v] != 1 or len(g[v]) != 1:
return False
else:
return True
# if node v is a 1-in-1-out node and is visited,
# it will be added in this set.
visited = set()
paths = []
for v in g:
# if v is not a 1-in-1-out node
if not oneone(v):
visited.add(v)
for w in g[v]:
non_branching_path = [v,w]
while oneone(w):
visited.add(w)
non_branching_path.append(g[w][0])
w = g[w][0]
paths.append(non_branching_path)
# check all isolated cycle
for v in g:
if oneone(v) and v not in visited:
visited.add(v)
cycle = [v]
w = g[v][0]
while oneone(w):
cycle.append(w)
visited.add(w)
if w == v:
break
w = g[w][0]
paths.append(cycle)
return pathsAssembling error-prone reads
Error-prone reads represent yet another barrier to real sequencing projects. Adding the single erroneous read CGTACGGACA (with a single error that misreads t as C) to the set of “broken” 5-mer reads from earlier in the lesson results in erroneous 5-mers CGTAC, GTACG, TACGG, ACGGA, and CGGAC after read breaking.
图11: bubble

This structure is called a bubble, which we define as two short disjoint paths (e.g., shorter than some threshold length) connecting the same pair of nodes in the de Bruijn graph.
Bubble removal occasionally removes the correct path, thus introducing errors rather than fixing them. To make matters worse, in a genome having inexact repeats, where the repeated regions differ by a single nucleotide or some other small variation, reads from the two repeat copies will also generate bubbles in the de Bruijn graph because one of the copies may appear to be an erroneous version of the other. Applying bubble removal to these regions introduces assembly errors by making repeats appear more similar than they are. Thus, modern genome assemblers attempt to distinguish bubbles caused by sequencing errors (which should be removed) from bubbles caused by variations (which should be retained).