NBT封面:纳米孔基因组测序快速临床诊断细菌性下呼吸道感染
文章目录
- Nanopore宏基因组测序快速临床诊断细菌性下呼吸道感染
- 热心肠日报
- 主要结果
- 新方法的开发
- 表1:常规医学培养结果和使用本流程宏基因组输出结果比对
- 宏基因组流程优化
- 图1 宏基因组流程示意图
- 确定检测极限
- 模拟微生物群落检测
- 优化检测流程
- 表2: 在去除人类DNA模式和未去除人类DNA模式下,对含有革兰氏阳性或革兰氏阴性病原菌样品中的人类DNA和细菌DNA进行定量。
- 抗生素抗性
- 表3: 基于优化的流程在ARMA中发现与病原菌生长相关的抗性基因。(41个样品,检测到183个抗性基因)
- 基于参考基因组组装
- 时间点分析
- 图2:细菌基因组组装、基覆盖率和抗生素基因检测
- 下面对图2分图展示和注解
- 方法
- 这整个流程翻译为中文,大家参考:
- 病原菌检测和抗生素抗性基因检测
- 细菌基因组组装
- 数据可用
- Reference
- 猜你喜欢
- 写在后面

Nanopore宏基因组测序快速临床诊断细菌性下呼吸道感染
Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection

译者:文涛 南京农业的大学
责编:刘永鑫 中科院遗传发育所
Nature Biotechnology [IF:31.864]
2019-06-24 Articles
DOI: https://doi.org/10.1038/s41587-019-0156-5
全文可开放获取 https://www.nature.com/articles/s41587-019-0156-5.pdf
第一作者:Themoula Charalampous1,8, Gemma L. Kay 1,2,8, Hollian Richardson1,8
通讯作者:Justin O’Grady 1,2*
其它作者:Alp Aydin2,
Rossella Baldan1,3, Christopher Jeanes4, Duncan Rae4, Sara Grundy4, Daniel J. Turner5
, John Wain1,2,
Richard M. Leggett6, David M. Livermore1,7
作者单位:
1 英国东安格利亚大学(
Bob Champion Research and Educational Building, University of East Anglia, Norwich Research Park, Norwich, UK)
2 英国诺里奇研究园区,生命科学学院(
Quadram Institute Bioscience,
Norwich Research Park, Norwich, UK)
热心肠日报
https://www.mr-gut.cn/papers/read/1074446025
Nature子刊:纳米孔宏基因组测序,6小时快速诊断下呼吸道感染病原体
创作:刘永鑫 审核:刘永鑫 08月02日
原标题:纳米孔宏基因组测序快速临床诊断细菌性下呼吸道感染
- 细菌性下呼吸道感染(LRI)培养法诊断敏感性差且太慢,不能指导早期的靶向抗生素治疗;
- 作者开发了一种用于细菌LRI诊断的宏基因组学方法,基于皂苷有效去除99.99%的宿主DNA并结合纳米孔测序;
- 该方法从样品到结果仅需6小时,对病原体检测的敏感性96.6%、特异性41.7%,同时可检测抗生素抗性基因;
- 结合定量PCR和特异性基因,特异性和灵敏度增加至100%。
- 纳米孔宏基因组学可以快速准确地表征细菌LRI,有助于减少广谱抗生素的使用。
主编评语:下呼吸道感染年导致300万人死亡且病原体类型广泛,目前基于培养的诊断需2~3天且敏感性较差。2019年7月Nature Biotechnology以《纳米孔上的临床宏基因组学》(Clinical metagenomics on a nanopore)为封面,刊登了英国东安格利亚大学Justin O’Grady博士及合作者共同发布的首个使用纳米孔技术的快速、经济的宏基因组测序方法,直接从患者呼吸道样本中准确快速地识别细菌病原体,并在6小时内准确检测抗性基因的突破性研究。
译者有话说:作为新一代测序技术的Nanopore最近一直博得人们的眼球,虽然目前其10%以上的错误率对于扩增子的运用存在比较大的问题,但是由于其超长的读长,实时测序,随断随测等优势在临床病原菌的检测方面具有很大优势,就这方面,本文的作者进行了尝试,通过开发了能去除宿主(人)DNA(去除率极高:99.99%)方法是本文进行Nanopore测序的基础,皂苷在这个过程中起着重要作用。以此为开端,作者优化提取DNA过程,测序建库过程,直到最后测序完成,使时间压缩到6个小时。并通过qPCR和抗生素抗性基因的共同验证,使得这一套体系在验证病人样本中达到了100%的特异性。作者也表示即将开展临床实验。
主要结果
新方法的开发
Pilot method development
使用我们开发的方法,对40名疑似细菌LRI患者的呼吸道样本进行了测试。该方法灵敏度为91.4%(计算网址:http://vassarstats.net/clin1.html ) 特异性100%。检测整个过程花费将近8个小时。通过qPCR证明了使用皂苷可以去除高达99.9%或~103倍的宿主DNA。微生物包括潜在的病原微生物通过WIMP流程分析鉴定。在测试的40个样品中有五个样品检测到了未通过常规培养方法检测到的其他病原菌:卡他莫拉菌 Moraxella catarrhalis (P8样品中); 大肠杆菌 Escherichia coli(P14样品中); 流感嗜血杆菌 H. influenzae(P22和P30样品中); 克雷伯氏肺炎菌 Klebsiella pneumoniae和 卡他莫拉菌 M. catarrhalis (P29样品中)(表1)。
在40个测试样品中的3个中未检测到使用常规临床微生物学培养方法检测到的病原微生物。这三个样本中两个都是混合感染(含有两种病原微生物),其中有一种病原菌没有检测到。另外,P3样本病原菌 pneumoniae ,P37样本中流感嗜血杆菌H. influenzae ,P34样本中金黄色葡萄球菌 Staphylococcus aureus也未检测到。
表1:常规医学培养结果和使用本流程宏基因组输出结果比对
Pilot metagenomic pipeline output compared with routine microbiology culture results

宏基因组流程优化
我们想通过改善病原菌细胞裂解程度来提高灵敏度(8.6%的假阴性。
首先在样品处理阶段通过球磨来提高裂解程度。
使用两个样品来优化流程,他们的病原菌分别为:金黄色葡萄球菌 S. aureus (革兰氏阳性) 和铜绿假单胞菌 P. aeruginosa (革兰氏阴性)。我们优化预处理过程并未改变病原菌为铜绿假单胞菌的样品DNA中的浓度;增加了病原菌为金黄色葡萄球菌的样品DNA中的浓度,两种预处理方法有所不同:多种酶混合液(enzyme cocktail)处理增加了四倍,球磨(bead-beating)增加了21倍。S. aureus病原菌在DNA中含量提高与金黄色葡萄球菌裂解程度增加有关(病原菌占总样本群落测序读长的80%)。所以我们采用了球磨(bead-beating)的方法改善DNA提取过程,去除用DNA酶处理的过程并相应的减少了干净DNA的步骤。在不影响效率的情况下,最终去除宿主DNA的提取过程从90分钟缩短为50分钟(附表 1a)。另外通过减少PCR延伸时间从6分钟缩短到4分钟,并通过验证发现6分钟和4分钟群落差异不大,平均读长略微缩短(<600 bp) (附表1b)。这些优化使得宏基因组文库整体制备时间较少了2.5小时,在DNA测序之前一共减少了4个小时。
图1 宏基因组流程示意图
Schematic representation of the metagenomic pipeline.

[外链图片转存失败(img-0Ietjrih-1566574453098)(http://210.75.224.110/Note/LiuYongXin/190624NBT/1b.png)]
一共需要6个小时完成整个流程(估计);加上采样和结果报告大约需要8个小时。
The turnaround time is approximately 6 h (optimized) and approximately 8 h (pilot) from sample collection to sample result. o/s: oscillations per second.
确定检测极限
Limit of detection
使用正常呼吸道菌群样品(normal respiratory flora)通过添加逐级稀释的金黄色葡萄球菌 S. aureus 和 大肠杆菌 E. coli。如果在 ≥ 1%的读长中存在,则每个重复被定义为’病原体’阳性(再WIMP流程中去除低质量序列)。
当微生物群落丰度较高时,大肠杆菌的最低检测限为105,金黄色葡萄球菌为104(附表2a)。当微生物群落整体丰度较低时,两种菌的检测限均为103(附表2b)。所以本方法可以检测限为103到105。但是基于不同的背景微生物群落,不同的取样人群最低检测限可能不同。
模拟微生物群落检测
Mock community detection
我们将病原菌加入到正常样品中(~103–106 c.f.u.),一式三份。检测去除人类DNA的方法是否会对细菌DNA造成损失。我们观察到大肠杆菌、流感嗜血杆菌、克雷伯氏肺炎菌、铜绿假单胞菌、金黄色葡萄球菌和嗜麦芽寡养单胞菌 Stenotrophomonas maltophilia 并没有细菌DNA的损失((ΔCq)) < 1)),而肺炎链球菌S. pneumoniae损失5.7倍(平均 ΔCq = 2.52)(附表3)。
优化检测流程
Optimized method testing
来自疑似LRI患者的41个呼吸道样品测试优化流程。通过qPCR,我们发现,未去除和去除人类DNA的样本之间人类DNA含量平均相差一万倍。通过优化的呼吸道病原菌检测优化灵敏度为96.6%,特异性为41.7%(Table 2)。从样本到结果共花费6个小时,其中纳米孔测序(MinION sequencing) 花费两个小时(Supplementary Table 4)。
使用我们优化后的方法检测到常规培养方法能检测到的病原菌和和八个样品中常规方法无法检测到的病原菌。这些常规方法无法检测的病原菌有*:K. pneumoniae* (S5样品), P. aeruginosa (S7样品), M. catarrhalis (S14和S39样品), S. pneumoniae(S8和S15样品), S. aureus (S29样品)S. pyogenes(S27样品) (表2)。有一些样品通过常规培养方法检测为正常的样本的,但是我们检测到至少两种潜在的致病菌,比如:S10 S21样本中H. influenzae 和 S. pneumoniae 病原菌; S11 S28 样本中的 S. pneumoniae 病原菌; S12样本中M. catarrhalis 和 H. influenzae 病原菌; S31 S32样本中 H. influenzae 和 E. coli病原菌。有一个常规培养方法为检测一种出病原菌的样本S9,使用我们优化的检测方法未检测出病原菌。这个样本为 P. aeruginosa 和 E. coli混合感染,并且只有E. coli 可以使用宏基因组测序检测到。通过常规培养方法检测S27,S38和S41样品为三种病原菌混合感染,并且使用优化的方法在所有三个样品中都检测到两种病原微生物。
这总共16个样品,要么遗漏病原菌或者检测出其他病原菌的样品,要么具有遗漏的病原体,基于这些问题我们使用qPCR优化。用作qPCR分析的DNA样本提取自未去除人类DNA的样本,防止我们的去除人类DNA方法对结果产生影响。首先通过宏基因组检测到的19个病原菌中有12个通过qPCR也检测到,这将我们优化的方法特异性提高到了83.3%。qPCR没有检测到S9号样品中的P. aeruginosa,将敏感性提高到了100%。
对具有有潜在致病菌的样本进行物种特异性基因的分析。该分析用于判断基于WIMP流程的是否错误分类。样品中包含大于1个 H. influenzae (siaT) 或者 S. pneumoniae (ply) 特异性基因片段就被认为存在这种致病菌。基于这种方法我们发现18个样品中有5个样本都检测到了H. influenzae/S. pneumoniae,虽然之前的流程和qPCR都没有检测到。与培养和qPRC的常规金标准相比,结合物种特异性基因的分析我们的检测方案的敏感性和特异性都优化到了100%。
表2: 在去除人类DNA模式和未去除人类DNA模式下,对含有革兰氏阳性或革兰氏阴性病原菌样品中的人类DNA和细菌DNA进行定量。
Human and bacterial DNA qPCR results for sputum samples infected by Gram-negative and Gram-positive bacteria with and without host nucleic acid depletion

备注:表格过长未显示完整,缺乏S24-S41样品。
抗生素抗性
Antibiotic resistance
根据常规检测结果,采用本文优化方法检测的样品几乎没有抗药性(附表 7)。测序鉴定了41个标本中的183个抗性基因。在183个抗性基因中,26个是固有的。剩下157个,其中24个与所见的表型相匹配。基于这些抗性基因结果,作者结合抗性测试,可以解释部分病原菌的抗生素抗性表型。但是仍然有9个抗性表型尚未通过测序得到的抗性基因解释,因此作者所开发的抗生素抗性基因检测方法的特异性和敏感性尚未确定,因为这需要对所有细菌进行分离和测序,工程量十分浩大。
表3: 基于优化的流程在ARMA中发现与病原菌生长相关的抗性基因。(41个样品,检测到183个抗性基因)
Resistance genes found by ARMA in relation to pathogens grown: optimized pipeline (41 samples; 183 genes detected)
[外链图片转存失败(img-RNHUxnyI-1566574453098)(http://210.75.224.110/Note/LiuYongXin/190624NBT/t3.png)]
基于参考基因组组装
Reference-based genome assembly
选择含有抗生素抗性病原菌的两个样品作为实例,直接从宏基因组数据基于参考的基因组进行组装。进行该分析以说明可以直接从呼吸道样品测得整个病原体基因组,用于公共健康和传染疾病控制。
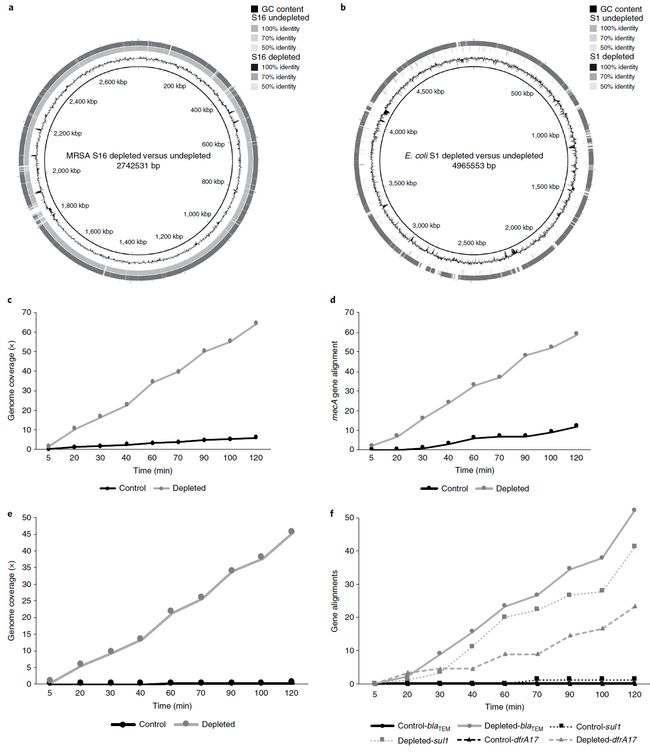
对 MRSA (S16)和 E. coli(对amoxicillin, co-amoxiclav 和co-trimoxazole具有抗性)进行组装。同时使用未去除人类DNA的样本作为对照同时测序并组装。使用两小时后和48小时后测序的结果进行组装。
就 MRSA 而言,使用两小时的测序结果,基于去除人类DNA优化的流程MRSA基因组覆盖度达到了47.9×并且组装了28个contigs(最长的片段479kbp,N50为400kbp),当测序时间延长到48个小时时基因组覆盖度达到了228.7×并且组装了22个contigs(最长的片段481kbp,N50为403kbp)。相反,未去除人类DNA的样本测序结果MRSA在两个小时后的组装覆盖度仅为3.9× 并且拼接成了69个contigs,48小时后覆盖度为17.5× 并且拼接成了33个contigs(图2a)。
对于具有抗生素抗性的E. coli,使用去除人类DNA的流程的两个小时后测序结果进行拼接,基因组覆盖度为33.5×,拼接成了83个contigs。使用48小时的测序结果,组装得到基因组覆盖度为增加165.7× ,共得到72个contigs( 最长的 contig = 474 kbp N50 = 178 kbp)。未去除人类DNA在两小时后只能组装出覆盖度为0.2×的基因组,并在48小时覆盖度仅仅提高到1.1× (图 2b)。
时间点分析
Time-point analysis
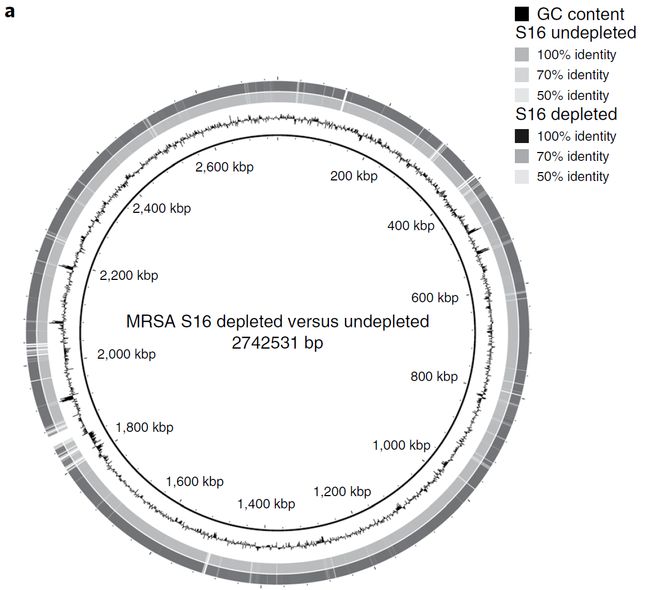
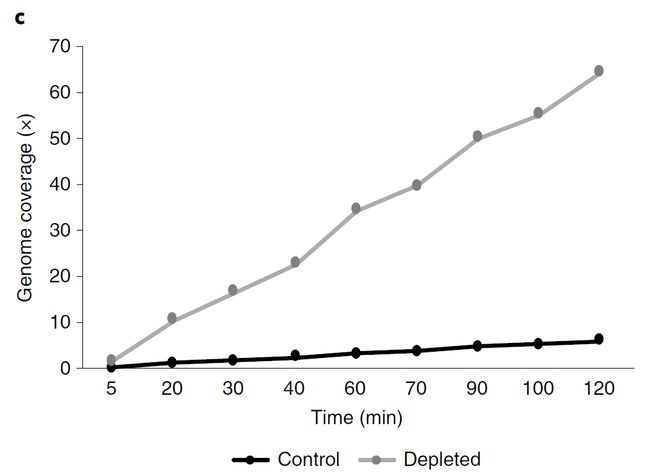
使用与基因组组装相同的样品,将来自测序的前2小时的数据按照时间序列进行比较,以突出去除宿主DNA对于优化时间的重要性。在测序的5分钟内,去除人类DNA的MRSA样品(S16)具有1.6×基因组覆盖度,而未去除的对照的覆盖度仅为0.2倍(图2c)。 5分钟后未去除人类DNA的样品中检测不到mecA基因,而在去除人类DNA的样品中在相同的时间点检测到两个mecA基因片段(图2d)。去除人类DNA的含E. coli样品(S1)在测序的20分钟内基因组覆盖率为5.7×,而未去除人类DNA的对照具有0.06×(图2e)。病原菌 E. coli 对amoxicillin (blaTEM gene), co-amoxiclav 和 co-trimoxazole (sul1 and dfrA17 genes)具有抗性。在测序两个小时后,在未去除人类DNA样品中未检测到blaTEM 和dfrA17 基因,只能检测到一个sul1基因片段。于此相对的是在去除人类DNA的样品中20分钟测序就可以检测到三种抗生素抗性基因,并且在两个小时后, 能检测到47 blaTEM基因, 37个sulf1基因 和 21个dfrA17基因(Fig. 2f)。
图2:细菌基因组组装、基覆盖率和抗生素基因检测
Fig. 2 | Bacterial genome assembly, genome coverage and antibiotic gene detection with depleted versus undepleted samples.
下面对图2分图展示和注解
图a:测序48小时后组装的抗甲氧西林金黄色葡萄球菌基因组草图;
a, MRSA after 48 h of sequencing.

图b:测序48小时后组装的大肠杆菌;
b, E. coli after 48 h of sequencing.
图c:在两个小时的测序过程中基于去除人类DNA和未去除人类DNA测序的MRSA基因组覆盖度
c, MRSA genome coverage of depleted versus undepleted during 2 h of sequencing.

图d:基于除人类DNA和未去除人类DNA的样品测序,mecA基因在两个小时测序过程数量变化。
d, mecA gene alignment of depleted versus undepleted during 2 h of sequencing.
图e:在两个小时的测序过程中基于去除人类DNA和未去除人类DNA测序的E. coli 基因组覆盖度
e, E. coli genome coverage of depleted versus undepleted during 2 h of sequencing.

图f:基于除人类DNA和未去除人类DNA测序的三个独立的样品,:三种基因在测序两个小时期间得到的序列数量变化。
f, blaTEM, sul1 and dfrA17 gene alignment of depleted versus undepleted during 2 h of sequencing.
方法
这整个流程翻译为中文,大家参考:

病原菌检测和抗生素抗性基因检测
Pathogen identification and antibiotic resistance gene detection
EPI2ME抗生素抗性分析流程(ONT, v.2.59.1896509)用于纳米孔测序原始序列的分析:这一流程用于鉴定样品微生物及其相关的抗生素抗性基因。WIMP流程可用于细菌,病毒,真菌,古细菌和人类序列及其呼吸道病原菌的鉴定。WIMP是一个基于Centrifuge和k-mer的序列鉴定工具,其基于Burrows–Wheeler变换和 Ferragina–Manzini指数,使用的RefSeq数据库。ARMA rev. 1.1.5 也包含了抗生素抗性分析流程。ARMA使用CARD数据可进行抗生素抗性基因识别和鉴定,这一目的是通过使用minimap2工具进行读长的对齐来实现的(准确度 > 75%,水平覆盖率> 40%)。在ONT网站上可以公开获得WIMP和ARMA流程的完整说明(https://nanoporetech.com/EPI2ME-amr)。NanoOK/NanoOK 也是公开使用的可以鉴定微生物和抗生素抗性基因的工具。输出的结果和WIMP和ARMA这两个管道类似。
对呼吸道宏基因组数据的初步分析表明,需要对序列定一个阈值来提高结果的准确性。常规来讲阈值在临床微生物实验室中用于一些感染案例,包括尿道和呼吸道的感染。用于呼吸道样本的临床阈值通常为每毫升105个病原菌(范围103-105 / ml,取决于样本类型)。在宏基因组测序样品中我们规定阈值为:相对丰度大于1%,并且q-score ≥ 20。我们选择大于这个阈值的序列进行后续分析,来忽略测序过程中出现的污染等。
细菌基因组组装
Bacterial genome assembly
首先使用Fast5-to-Fastq进行基因组装配,以去除短于2,000bp并且平均质量得分低于7的序列(https://github.com/rrwick/Fast5-to-Fastq)。 使用Porechop去除在序列中间或者两端的barcode。完成后重新识别每个样品上的标签(https://github.com/rrwick/Porechop)。使用minimap2工具(默认参数)将过滤后(通过WIMP流程过滤得到病原菌的序列)的序列根据参考基因组比对。Canu工具用来对序列进行组装和矫正组装成重叠群。BLAST Ring Image Generator (BRIG) 用于比对基因组的组装结果。
数据可用
Data availability
所有原始序列和组装结果存于欧洲核酸数据库。项目号为:PRJEB30781。
Reference
Charalampous, T. et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nature Biotechnology 37, 783-792, doi:10.1038/s41587-019-0156-5 (2019).
新闻稿 NBT封面:纳米孔宏基因组6小时识别下呼吸道病原体
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA