Nature:基于宏基因组测序构建人类肠道微生物组参考基因集
文章目录
- 基于宏基因组测序构建人类肠道微生物组参考基因集
- 文章影响
- 作者简介
- 热心肠日报
- 摘要
- 正文
- 宏基因组测序肠道微生物组
- 图1. 人类肠道微生物组的覆盖度
- 人类肠道微生物组的基因集
- 图2. 预测人体肠道微生物组中的ORF
- 常见核心细菌
- 图3. 57种高频微生物基因组在个体中的相对丰度的分布
- 图4. IBD病人和健康个体细菌物种丰度的差异
- 流行基因集编码的功能
- 细菌功能对肠道的生命至关重要
- 图5:含有枯草芽孢杆菌必需基因的簇
- 图6:最小肠道基因组和宏基因组的特征
- 基因组和宏基因组的功能互补
- 讨论
- 猜你喜欢
- 写在后面

基于宏基因组测序构建人类肠道微生物组参考基因集
A human gut microbial gene catalogue established by metagenomic sequencing
Nature [IF:43.07]
04 March 2010 Article
DOI: https://doi.org/10.1038/nature08821
第一作者:Junjie Qin(覃俊杰)1
,Ruiqiang Li(李瑞强)1
通讯作者:S. Dusko Ehrlich ([email protected])6
& Jun Wang (王俊)([email protected])1,13
其它作者:Jeroen Raes, Manimozhiyan Arumugam, Kristoffer Solvsten Burgdorf, Chaysavanh Manichanh, Trine Nielsen, Nicolas Pons, Florence Levenez, Takuji Yamada, Daniel R. Mende, Junhua Li, Junming Xu, Shaochuan Li, Dongfang Li, Jianjun Cao, Bo Wang, Huiqing Liang, Huisong Zheng, Yinlong Xie, Julien Tap, Patricia Lepage, Marcelo Bertalan, Jean-Michel Batto, Torben Hansen, Denis Le Paslier, Allan Linneberg, H. Bjørn Nielsen, Eric Pelletier, Pierre Renault, Thomas Sicheritz-Ponten, Keith Turner, Hongmei Zhu, Chang Yu, Shengting Li, Min Jian, Yan Zhou, Yingrui Li, Xiuqing Zhang, Songgang Li, Nan Qin, Huanming Yang, Jian Wang, Søren Brunak, Joel Doré, Francisco Guarner, Karsten Kristiansen, Oluf Pedersen, Julian Parkhill, Jean Weissenbach, H. I. T. Consortium Meta, Maria Antolin, François Artiguenave, Hervé Blottiere, Natalia Borruel, Thomas Bruls, Francesc Casellas, Christian Chervaux, Antonella Cultrone, Christine Delorme, Gérard Denariaz, Rozenn Dervyn, Miguel Forte, Carsten Friss, Maarten van de Guchte, Eric Guedon, Florence Haimet, Alexandre Jamet, Catherine Juste, Ghalia Kaci, Michiel Kleerebezem, Jan Knol, Michel Kristensen, Severine Layec, Karine Le Roux, Marion Leclerc, Emmanuelle Maguin, Raquel Melo Minardi, Raish Oozeer, Maria Rescigno, Nicolas Sanchez, Sebastian Tims, Toni Torrejon, Encarna Varela, Willem de Vos, Yohanan Winogradsky, Erwin Zoetendal, Peer Bork
作者单位:
1 中国深圳,华大基因(BGI-Shenzhen, Shenzhen 518083, China)
6 法国乔伊·乔萨斯,法国国立农学研究所(Institut National de la Recherche Agronomique, 78350
Jouy en Josas, France.)
文章影响
该文章发表9年,引用超7000次,是2010年Nautre最高引文论文,在微生物组领域中,相当于人类基因组计划一般的影响力。
作者简介
9年后,两位一作和最后一位通讯,均成为本领域的顶级大佬,每个人都身价十亿起!看下文简介。

- 覃俊杰
中科院北京基因组研究所博士毕业,原华大基因研究院微生物方向第一负责人、从2006年开始,从事基于二代测序技术的微生物基因组、宏基因组研究和技术转化。完成的重要项目有:欧洲人肠道菌群宏基因组图谱的构建、2011年德国致病大肠杆菌的快速基因组解析、中国人2型糖尿病与肠道菌群的宏基因组关联分析、欧洲人肥胖与肠道菌群的关联分析、临床特定疾病的菌群组成与功能研究等。在宏基因组学领域开发了许多重要的技术工具与方法。其中,肠道菌群宏基因图谱构建的文章作为2010年3月的《Nature》杂志封面,迄今为止已经得到7000多次引用,是文章发表当年(2010年)全球引用率最高的生物学论文。 2014年7月成立谱元科技,http://www.promegene.com/ http://microbiota.cn/ ,主营方向为微生物检测和相关产品,2016年2月估值2亿完成首轮融资。
- 李瑞强
1979年出生于中国江苏。本科就读于东南大学应用物理学专业,2002年本科毕业即加入了华大基因,从事基因组与生物信息学研究。期间于2004年在英国Sanger 基因组中心访问学习,于2010年获得丹麦哥本哈根大学生物学博士学位。曾任北京大学研究员。
2011年成立诺禾致源(http://www.novogene.com),2015年科技服务的国内市场收入达到3.5 亿,超越华大基因,成为业内最大的科研服务提供商。2016年11月5亿元的B轮融资,估值有50亿以上;最近正在申请创业板上市中。
- 王俊
1976年出生,97年北大本科,北大博士。国家杰出青年基金获得者,973首席科学家;曾任华大基因CEO,华大基因研究院院长;丹麦哥本哈根大学、香港大学客座教授。发表三百余篇论文,第一作者篇或通讯作者百余篇。其中在Science、Nature系列杂志、Cell及NEJM上发表百余篇(20篇为封面文章)。2015年创立碳云智能 https://www.icarbonx.com/about.html ,成立仅17个月即获腾讯10亿投资,估值超10亿美元,智能穿戴设备,营养、运动、护肤,免疫组学检测。
热心肠日报
第148期:花费近10亿RMB,两大微生物组计划有何成果?(经典回顾)
http://www.mr-gut.cn/daily/show/1270648224
160908话题:系统回顾人类微生物组计划和人类肠道宏基因组计划的经典文献。
Nature:在人类肠道中鉴定出330万个微生物基因(2010)
http://www.mr-gut.cn/papers/read/1083232040
创作:赵弘烨 Bob 审核:蓝灿辉 | 热心肠先生12月30日
①作为人肠道宏基因组(MetaHIT)计划的一部分,研究者使用Illumina测序仪,组装并鉴定了124个欧洲人的粪便标本中的330万个不重复的微生物基因;
②这些超过人类基因150倍的基因集包含了绝大多数人类的主要肠道微生物基因,并且大部分基因在人群中共有;
③细菌基因比例超过99%,这群人中有1100种左右种细菌,其中160个优势物种为人群所共有;
④作者还对肠道宏基因组和肠细菌基因组进行了功能性分析。
主编推荐语:
经典回顾,这是MetaHIT抢在HMP团队之前在Nature上发出的第一个重量级阶段性研究成果,也是覃俊杰博士的成名之作,迄今被引用次数已近4000次。
摘要
要了解肠道微生物对人类健康的影响,评估其遗传潜力至关重要。在这里,我们描述了基于Illumina的宏基因组测序,并组装和表征了330万个非冗余微生物基因,这些基因来自124个欧洲个体的粪便样本获得的576.7 GB 序列。该基因组比人类基因数量大约150倍,其中包含该队列中绝大多的微生物基因,并且可能包括大部分人类肠道微生物基因。这些基因在该队列的个体之间很大程度上共享。超过99%的基因是细菌,这表明整个队列中共有1000至1150种流行细菌,每个个体至少有160种这样的细菌,它们在很大程度上也是共享的。我们分别根据所有个体和大多数细菌的功能定义描述了最小的肠道宏基因组和最小的肠道细菌基因组。
正文
据估计,我们体内的微生物共同构成多达100万亿个细胞,是人类细胞数量的十倍(这是历史数据,最新Cell统计结果为1倍,详见《Cell:人体肠道细菌与自身细胞的比例究竟是多少?》),并暗示它们编码的独特基因比我们自己的基因组多100倍(这可以作为我们突出宏基因组重要性的证据)。大多数微生物都生活在肠道中,对人类的生理和营养产生深远的影响,对人类的生命至关重要[2,3]。此外,肠道微生物有助于从食物中获取能量,肠道微生物组的变化可能与肠道疾病或肥胖有关[4,5,6,7,8]。
为了理解和利用肠道微生物对人类健康的影响,有必要对肠道微生物群落的内容,多样性和功能进行解读。基于16S核糖体RNA基因(rRNA)测序的方法显示两个细菌分支,即拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes),构成了已知系统发育类别的90%以上,并主导着远端肠道菌群。研究还表明,健康个体之间肠道微生物组的多样性差异很大。尽管这种差异在婴儿中尤为明显,但在生命的后期,肠道微生物组会聚成更相似的门。
宏基因组测序是分析复杂微生物群落中常用的rRNA基因测序的有力替代方法。在人类肠道中进行的此类研究已经从美国或日本的33个个体的粪便样本中产生了约3 Gb的微生物序列(Gb)。为了更全面地了解人类肠道微生物基因,我们使用了Illumina基因组分析仪(GA)技术对来自124个欧洲成年人的粪便样本中的总DNA进行了深度测序。我们产生了576.7 Gb的序列,几乎是所有先前研究的200倍,将其组装成重叠群,并预测了330万个独特的开放阅读框(ORF)。该基因集实际上包含了我们队列中所有流行的肠道微生物基因,提供了对肠道细菌生命至关重要功能的广泛描述,并表明许多细菌物种是由不同个体共享的。我们的结果还表明,短读长宏基因组测序可用于对生态复杂环境的遗传潜力进行全面表征。
宏基因组测序肠道微生物组
Metagenomic sequencing of gut microbiomes
作为MetaHIT(Metagenomics of the Human Intestinal Tract,人体肠道宏基因组学)项目的一部分,我们收集了来自丹麦和西班牙的124名健康,超重和肥胖的成年个体成年人以及炎症性肠病(IBD)患者的粪便标本(补充表1) 。从粪便标本中提取总DNA,每个样品平均产生4.5 Gb(2到7.3Gb)的序列,这使我们能够捕获大部分新颖性(参见方法和补充表2)。总共,我们获得了576.7 Gb的序列(补充表3)。
为了从人类肠道中产生广泛的微生物基因集,我们首先将短的Illumina读长组装成更长的重叠群,然后可以通过标准方法对其进行分析和注释。使用SOAPdenovo,这是一种基于de Bruijn图的工具,专门用于组装非常短的长,我们对所有Illumina GA序列数据进行了从头组装。因为期望个体之间的高度多样性,所以我们首先独立地组装每个样本(补充图3)。多达42.7%的Illumina GA读段被组装成总共658万个重叠群,其长度 > 500 bp,重叠群的总长度为10.3 Gb,N50长度为2.2 kb(补充图4),范围为12.3至237.6 Mb(补充表4)。来自任何一个样品的几乎35%的读长可以被映射到来自其他样品的重叠群,表明存在公共序列核心。
为了评估基于Illumina GA的组装质量,我们将样本MH0006和MH0012的重叠群映射到来自相同样本的Sanger读长(补充表2)。映射到至少一个Sanger读长的重叠群中,共有98.7%的共线性超过了所映射区域的99.6%。这与两个样品之一(MH0006)的454测序所产生的重叠群相似,其中97.9%在99.5%的比对区域内共线。我们估计,基于Illumina和454的重叠群的组装错误分别为14.2和 20.7 个每兆碱基(megabase, Mb)(请参见“方法”和“补充图5”),表明基于短读长和长读长的组装具有可比的精度。
为了完成重叠群的设置,我们合并了来自所有124个样本的未组装读长,并重复了从头组装过程。这样就产生了大约40万个重叠群,长度为370 Mb,N50长度为939 bp。因此,我们最终重叠群的总长度为10.7 Gb。在Illumina GA序列的576.7 Gb序列中,约有80%可以以90%相似度的阈值与重叠群进行比对,从而可以适应肠道中的测序错误和菌株变异性(图1),几乎是序列42.7%的两倍。 SOAPdenovo将其组装成重叠群,因为组装使用了更严格的标准。这表明,Illumina序列的绝大多数由我们的重叠群代表。
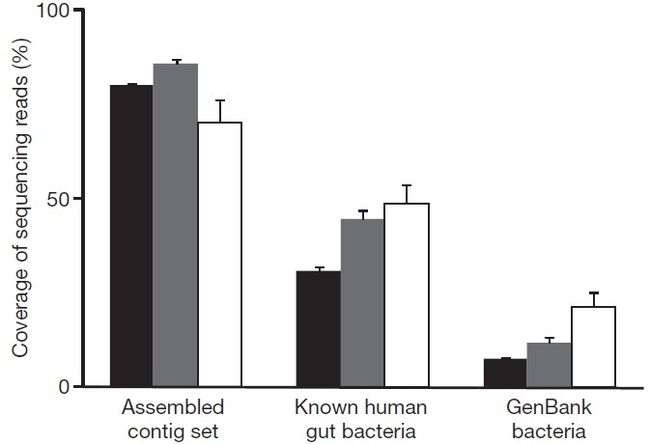
图1. 人类肠道微生物组的覆盖度
Figure 1: Coverage of human gut microbiome.
使用三种人类微生物测序读长:来自本研究的124个人的Illumina GA读长(黑色; n = 124),来自18对人类双胞胎及其母亲的Roche 454读长(灰色; n = 18),自13个日本人个体的Sanger读长(白色; n = 13);将它们与每个参考序列组比对。 绘制平均值±s.e.m。
为了将人类肠道微生物组在我们的重叠群中的代表性与之前的工作进行比较,我们将其与两个最大的已发表的肠道宏基因组研究的读长(来自18位美国成年人罗氏454测序读长1.83 Gb,和从13名日本成年人和婴儿中Sanger读长0.79 Gb,使用90%的相似度阈值进行比较。从日本和美国样品中分别获得的总读长的70.1%和85.9%可以与我们的重叠群进行比对(图1),这表明重叠群包含了先前研究的很大一部分序列。相比之下,分别来自日本和美国样本的读数未涵盖我们重叠群的85.7%和69.5%,这突出了我们捕获的新颖性。
前两项研究和本研究中只有31.0–48.8%的读长可与194个公共人类肠道细菌基因组进行比对(补充表5),而与GenBank中保存的细菌基因组相比则为7.6–21.2%(图1)。 。这表明通过对分离的细菌菌株的基因组进行测序获得的参考基因组仍然规模有限。
人类肠道微生物组的基因集
A gene catalogue of the human gut microbiome
为了建立非冗余的人类肠道微生物组基因组,我们首先使用MetaGene程序来预测重叠群中的ORF,并发现了14,048,045个长度大于100 bp的ORF(补充表6)。它们占据了重叠群的86.7%,与完全测序的基因组的发现值相当(〜86%)。三分之二的ORF似乎不完整,可能是由于我们的重叠群大小(N50为2.2 kb)所致。接下来,我们通过成对比较,使用非常严格的标准,即95%的相似度超过较短的ORF长度的90%,删除了多余的ORF,这可以融合直系同源物,也可以避免由于可能的测序错误而导致数据集膨胀(请参见方法,构建非冗余基因集)。但是,最终的非冗余基因集包含多达3,299,822个ORF,平均长度为704 bp(补充表7)。
我们定义非冗余基因集中的“流行基因(prevalent genes)”,因为它们是在最丰富的读长组装而成的重叠群上编码的(请参见“方法”)。根据非冗余基因的最小序列覆盖率(接近3倍)和每个个体产生的总Illumina序列长度(平均4.5 Gb)估计,流行基因的最小相对丰度为 ~ 6 × 10-7。假设平均基因长度为 0.85 kb(即 3 × 0.85 × 10-3 / 4.5×10-9)。
我们将330万个肠道ORF映射到人类肠道中89个常见参考微生物基因组的319,812个基因(目标基因)。在90%的同一性阈值下,80%的靶基因的长度至少有80%被单个肠道ORF覆盖(图2b)。这表明该基因组包括大多数已知的人类肠道细菌基因。

图2. 预测人体肠道微生物组中的ORF
Figure 2: Predicted ORFs in the human gut microbiome.
a,非冗余基因的数目与测序样本量的函数。基因积累曲线对应于Sobs(Mao Tau)值(观察到的基因数),该值是使用EstimateS(版本8.2.0)对随机选择的100个样本(由于内存限制)计算得出的。
b,来自89种常见肠道微生物物种的基因覆盖数量和比例的关系,采用三种不同相似度(补充表12)。
c,基于已知(特征明确的)直系同源基团(OG;底部),已知加未知直系同源基团(包括例如假定的、预测的、保守的假定功能;中间)和从宏基因组中恢复直系同源的基因,通过调查的样本数量捕获的功能数组和新基因家族(> 20个蛋白质)(上)。箱线表示第一个和第三个四分位数(分别为第25个和第75个百分位数)之间的四分位间距(IQR),内部的线表示中位数。晶须分别表示距第一个和第三个四分位数的1.5倍IQR内的最低和最高值。圆圈表示晶须以外的异常值。
我们检查了在所有个体中发现的流行基因的数量,该数量是测序样本量的函数,需要至少两个支持性读长基因调用recall(图2a)。基于指示的覆盖范围丰富度估计值(coverage richness estimato, ICE),是由100个人确定的(EvaluateS程序可以容纳的最高人数),表明我们的目录涵盖了85.3%的流行基因。尽管这可能被低估了,但它仍然表明该目录包含了该队列的绝大多数流行基因。
每个人携带536,112±12,167(平均值±s.e.m.)流行基因(补充图6b),表明330万个基因库中的大多数一定是共享的。但是,大多数流行基因仅在少数个体中发现:2,375,655存在于不到20%的个体,仅294,110个基因在50%的个体中发现(我们称这些“常见”基因)。这些值取决于采样深度。 MH0006和MH0012的测序揭示了更多的目录基因,以低丰度存在(补充图7)。然而,即使在我们常规的采样深度下,每个人也拥有204,056±3,603(均值±±s.e.m。)共同基因,这表明一个人的总基因库中大约有38%是共享的。有趣的是,IBD患者平均拥有的基因比非IBD患者少25%(补充图8),这与前者的细菌多样性低于后者的观察结果一致。
常见核心细菌
Common bacterial core
深度宏基因组测序提供了探索队列中一组常见微生物物种(共有核心)存在的机会。为此,我们使用了650个测序细菌和古细菌基因组的非冗余集(请参见方法)。我们使用90%的同一性阈值将每个人类肠道微生物样品的Illumina GA读长比对到基因组集上,并确定了仅对准集合中单个位置的读数所覆盖的基因组比例。在1%的覆盖率下,典型的肠道细菌基因组平均长度约为40kb,比通常用于物种鉴定的16S基因的长度长25倍,我们在所有个体中检测到18种,其中57种 ≥ 90%和75 ≥ 50%的个人中存在(补充表8)。当覆盖率达到10%时,要求样品的丰度提高到10倍,我们仍然在 ≥ 90%的个体中发现了13种上述物种,而在 ≥ 50%的个体中发现了35种。
当样品MH0006和MH0012的累积序列长度分别从3.96 Gb增加到8.74 Gb和从4.41 Gb增加到11.6 Gb时,在1%覆盖率阈值下,两者共有的菌株数从135个增加了25%。 这表明存在一个比我们在每个人常规使用的序列深度处观察到的更大的核心微生物组。
个体中微生物种类丰富的变异性会极大地影响共同核心的识别。为了可视化这种可变性,我们比较了我们队列中个体与不同基因组比对的测序读长的数量。即使对于90%的个体中基因组覆盖率> 1%的个体中最常见的57种(补充表8),个体间变异性也在12到2187倍之间(图3)。不出所料,拟杆菌门和硬壁菌门的丰度最高。

图3. 57种高频微生物基因组在个体中的相对丰度的分布
Figure 3: Relative abundance of 57 frequent microbial genomes among individuals of the cohort.
基于对网络的分析得出了一个复杂的物种相关性模式,其特征是在属和科上都具有簇。基于至少覆盖率≥1%的物种的成对皮尔逊相关系数,进行网络分析(补充图)。 9)突出的簇包括一些最丰富的肠道菌种,例如拟杆菌属和 Dorea / Eubacterium / Ruminococcus组的成员,还有双歧杆菌,变形菌门和链球菌/乳杆菌组。这些结果 表明,出于尚待确定的原因,我们队列中的不同个体中可能存在相似的细菌群。
以上结果表明,基于Illumina的细菌谱分析应揭示健康个体与患者之间的差异。为了验证这一假设,我们比较了IBD患者和健康对照者(补充表1),因为先前已报道两者具有不同的微生物群。基于相同155种物种的主成分分析清楚地将患者与健康个体分开,并将溃疡性结肠炎与克罗恩病患者分开(图4),证实了我们的假设。

图4. IBD病人和健康个体细菌物种丰度的差异
Figure 4: Bacterial species abundance differentiates IBD patients and healthy individuals
基于健康状况作为基础变量的主成分分析,是基于对至少1例队列中Illumina读长的155种≥1%基因组覆盖率的物种进行的,对14例健康个体和25例IBD患者(21例溃疡性 结肠炎和4种克罗恩氏病)(来自西班牙)(补充表1)。 绘制了两个主成分(PC1和PC2),它们代表整个差异的7.3%。 聚类(以点表示)的个人,并为每个类别计算重心; 使用蒙特卡洛检验(999个重复)评估了健康状况和物种丰富度之间的显著性的P值。
流行基因集编码的功能
Functions encoded by the prevalent gene set
我们通过将预测基因与非冗余蛋白序列的整合NCBI-NR数据库、KEGG(基因和基因组京都百科全书)通路中的基因、以及COG(直系同源群)和eggNOG数据库进行比对,对预测的基因进行了分类。分别有77.1%的基因分类为系统发育类型(phylotypes),eggNOG注释比例为57.5%,KEGG注释比例为47.0%,KEGG通路基因为18.7%(补充表9)。系统发育分配的基因中,几乎所有基因(99.96%)都属于细菌和古细菌,反映了它们在肠道中的优势(详者注:真核生物的基因没有很好的注释,导致此结果偏高)。未映射到直系同源基因的基因被聚类为基因家族(请参见方法)。为了研究流行基因集的功能内容,我们计算了n个个体(n = 2–124;见图2c)的任何组合中存在的直系同源基团和/或基因家族的总数。这种稀疏性分析表明,“已知”功能(在eggNOG或KEGG中注释)迅速饱和(观察到5569组值):对50个个体的任何子集进行采样时,大多数被检测到。然而,四分之三的普遍肠道功能由未表征的直系同源基团和/或全新的基因家族组成(图2c)。当包括这些组时,稀疏度曲线仅在最后阶段才开始趋于平稳,并达到更高的水平(检测到19,338组),这证实了大量个体的大量采样对于捕获如此大量的新颖性/功能未知。
细菌功能对肠道的生命至关重要
Bacterial functions important for life in the gut
来自人类肠道的细菌基因的广泛的非冗余基因集提供了一个可以识别对于这种环境下的生命至关重要的细菌功能的机会。细菌在肠道环境中具有旺盛的功能(即“最小肠道基因组”),而涉及整个生态系统稳态的功能则是跨多种物种编码的(“最小肠道宏基因组”)。预计第一组功能将存在于大多数或所有肠道细菌物种中,第二组是大多数或所有个体的肠道样本。
为了确定由最小的肠道基因组编码的功能,我们使用以下条件:它们应该存在于大多数或所有肠道细菌物种中,因此在基因目录中的出现频率高于某些肠道细菌物种中存在的功能频率。在对基因长度和拷贝数进行归一化之后,可以从归类到不同eggNOG簇的基因数量中推导出不同功能的相对频率(补充图10a,b)。我们通过基因频率对所有簇进行了排序,并确定了包括指定众所周知基本细菌功能簇的范围,例如针对经过深入研究的硬毛枯草芽孢杆菌实验确定的那些,假设该范围内的其他簇同样重要。不出所料,包括大多数枯草芽孢杆菌必需簇(86%)的范围位于排名的最顶端(图5)。具有大肠杆菌28个必需基因的簇中约有76%在此范围内,证实了我们方法的有效性。这表明在该范围内发现了1,244个宏基因组簇(补充表10;以下称为“范围簇”),它们对肠道中的生命至关重要。

图5:含有枯草芽孢杆菌必需基因的簇
Figure 5: Clusters that contain the B. subtilis essential genes.
通过簇包含的基因数量对簇进行排序,并通过平均长度和拷贝数对其进行归一化(参见补充图10),并针对连续的100个簇组确定具有基本枯草芽孢杆菌基因簇的比例。 范围表示包含86%枯草芽孢杆菌必需基因的簇分布部分。
我们在范围簇中发现了两种类型的功能:所有细菌都需要这些功能(管家)和潜在地针对肠道的功能。 在第一类的许多例子中,有一些功能是主要的代谢途径(例如,中央碳代谢,氨基酸合成)和重要的蛋白质复合物(RNA和DNA聚合酶,ATP合酶,一般分泌细胞质)。 毫不奇怪,范围簇在KEGG代谢途径上的投影给出了整体肠道细胞代谢高度整合的视角(图6a)。

图6:最小肠道基因组和宏基因组的特征
Figure 6: Characterization of the minimal gut genome and metagenome.
a,使用iPath工具将最小的肠道基因组投影到KEGG途径上。
b,最小肠道基因组和宏基因组的功能组成。稀有且频率地指的是测序的eggNOG基因组中存在。
c,最小肠宏基因组大小的估计。显示了已知的直系同源基团(红色),末知的直系同源基团(蓝色)和直系同源基团加上新的基因家族(> 20个蛋白质;灰色)(方框图和晶须图的定义见图2c)。插图显示了肠道最小微生物组的组成。大圆圈:根据STRING细菌基因组中直系同源基团的出现,将最小基因组分类。常见(25%),罕见(35%)和稀有(45%)是指分别存在于STRING细菌基因组中的 > 50%,<50%但 > 10%和<10%的功能。小圆圈:稀有直系群的组成。未知(80%)没有注释或特征不清,而已知细菌(19%)和噬菌体相关(1%)直系同源基团具有功能描述。
推测的肠道特异性功能包括那些与宿主蛋白(胶原蛋白,纤维蛋白原,纤连蛋白)的粘附或与收集在血液和上皮细胞上的globoseries糖脂的糖有关的功能。此外,15%的范围簇编码的功能存在于< 10%的eggNOG基因组中(参见补充图11),并且很大程度上(74.3%)未定义(图6b)。对这些细菌的详细研究应导致对肠道细菌生命活动更深刻的理解。
为了确定由最小肠道宏基因组编码的功能,我们计算了该队列中的个体共享的直系同源基团。这个最小的集合有6,313个功能,比以前的研究估计的要大得多。只有2,069个带功能注释的直系同源基团,表明它们严重低估了个体之间通用功能互补序列的真实大小(图6c)。最小的肠道宏基因组包括相当一部分功能(〜45%),这些功能存在于<10%的测序细菌基因组中(图6c,插图)。在肠道生态系统中,可能需要在124个个体中发现的这些原本稀有的功能。这些直系同源基因组中有80%的基因只具有较差的功能描述,这突显了我们对肠道功能的了解有限。
在已知部分中,约5%编码(原)噬菌体相关蛋白,这表明噬菌体在肠道稳态中普遍存在并可能具有重要的生态作用。似乎对于最小的基因组至关重要的最惊人的次级代谢与从宿主饮食和/或肠内壁中收获的复合糖和聚糖的生物降解无关,这并非出乎意料。实例包括果胶(及其单体,鼠李糖)和山梨糖醇,水果和蔬菜中普遍存在但未被人类吸收或吸收不良的糖类的降解和摄取途径。由于发现某些肠道微生物会同时降解它们,因此这种能力似乎被肠道生态系统选择为非竞争性能源。除此之外,发酵能力(例如甘露糖,果糖,纤维素和蔗糖)也是最小基因组的一部分。这些共同强调了肠道生态系统对糖复降解功能的强烈依赖性。
基因组和宏基因组的功能互补
Functional complementarities of the genome and metagenome
肠道宏基因组与人类基因组之间互补性的详细分析超出了本研究的范围。为了提供概述,我们考虑了两个因素:最小基因组中功能的保守性和一个或另一个中功能的存在/不存在(补充表11)。肠道细菌主要利用发酵来产生能量,将糖部分转化为短链脂肪酸,宿主将其用作能源。乙酸盐对肌肉,心脏和脑细胞很重要,丙酸盐可用于宿主肝的新糖原形成过程,而丁酸盐对肠上皮细胞也很重要。除短链脂肪酸外,许多氨基酸对于人类是必不可少的,并且可以由细菌提供。同样,细菌可以为宿主贡献某些维生素(例如,生物素,叶醌)。这些分子生物合成的所有步骤均由最小基因组编码。
肠道细菌似乎能够降解多种异生质,包括未修饰的和卤化的芳香族化合物(补充表11),即使大多数途径的步骤都不是最小基因组的一部分,并且仅在一部分个体中发现。一个特别有趣的例子是苯甲酸盐,这是一种常见的食品补充剂为E211。它被最小的基因组编码的辅酶A连接途径降解,生成了生物素的前体庚二酰辅酶A,表明该食品补充剂可能对人类健康具有潜在的有益作用。
讨论
我们已使用来自欧洲(北欧和地中海)起源的124个队列的全部粪便DNA的大量Illumina GA短读长测序,来建立非冗余人类肠道微生物基因目录。该目录包含330万个微生物基因,比人类基因多150倍,并且包括我们队列中绝大多数(> 86%)流行基因。该目录可能包含人类人口中绝大多数的肠道微生物基因,其原因如下:(1)可以对包括美国和日本人在内的三项先前研究的超过70%的宏基因组读物进行定; (2)我们的研究组中存在来自89个常见肠道参考基因组的微生物基因中的约80%。该结果代表了原理的证明,即短读测序可用于表征复杂的微生物组。
在我们的工作中没有对每个个体的完整细菌基因进行采样。然而,在我们这个队列中携带的330万个基因中,我们每个都检测到了536,000个流行的独特基因。不可避免地,个体在很大程度上共享公共库的基因。在目前的测序深度中,我们发现来自每个个体的基因几乎有40%与该队列的至少一半个体共享。国际人类微生物组联盟(International Human Microbiome Consortium)计划在未来进行全球范围的研究,必要时将完善我们的基因目录并确定共享基因比例的界限。
基本上,我们目录中的所有基因(99.1%)都是细菌起源的,其余大部分是古细菌,只有0.1%的真核和病毒起源。因此,该基因目录相当于具有平均大小的基因组的约1,000种细菌的基因目录,编码约3,364个非冗余基因。我们估计目录中可能缺少不超过15%的我们流行的基因,并且表明该人群中所包括细菌的数量不超过1,150种,足以被我们的抽样检测到。考虑到在本研究和以前的研究中微生物序列之间存在很大的重叠,我们建议丰富的肠道细菌种类的数量可能不会比我们队列中观察到的数量高很多。根据平均流行基因数量估算,我们队列中的每个人至少拥有160种此类细菌,因此必须共享许多细菌。
我们将大约12%的参考集基因(404,000)分配给194个已测序的肠道细菌基因组,并因此可以将它们与细菌种类相关联。通过人类微生物组计划和MetaHIT,预计国际人类微生物组联盟将对至少1,000个与人类相关的细菌基因组进行测序。这与我们队列中的优势物种数量相对应,并且有望在人类肠道中得到更广泛的应用,并且应该能够使更广泛的基因分配给物种。但是,我们使用了目前可用的测序基因组,进一步探讨了我们队列中广泛共享的物种的概念,并确定了> 50%的个体共有的75种和> 90%的共有57种。这些数量可能会随着测序参考菌株的数量和更深层的采样而增加。确实,测序深度增加了2到3倍,使我们可以检测到的两个个体之间共享的物种数量增加了25%。大量共有物种支持以下观点,即普遍存在的人类微生物组的大小有限且不会太大。
以前的大多数研究都使用16S RNA基因进行研究,因此如何将这种观点与肠道中无数细菌种类的相当大的人际差异相一致?这些研究的抽样深度可能不足以揭示低丰度时常见的物种,并强调相对少数优势物种组成的差异。我们发现我们队列中的57个最常见物种的丰度变异性非常高(12至2,200倍)。但是,最近一项基于16S rRNA的研究得出结论,存在至少50%受研究个体共有的常见细菌“核心” 。
将来,我们将在正在进行的MetaHIT临床研究的背景下,对我们队列中各个个体的细菌基因进行详细比较。基因家族的聚类使我们能够捕获流行基因集的几乎全部功能潜力,并揭示相当大的新颖性,与以前的工作相比,功能类别扩展了约30%。同样,该分析揭示了该队列中每个个体均保守的功能核心,该核心反映了完整的最小人类肠道宏基因组,该基因组编码于许多物种中,可能是肠道生态系统正常运行所必需的。这个最小的基因组大小超过了先前报道的核心基因组的数倍。它包括已知对宿主与细菌相互作用很重要的功能,例如复杂多糖的降解,短链脂肪酸的合成,必不可少的氨基酸和维生素。最后,我们还确定了归因于最小肠道细菌基因组的功能,这是任何细菌在该生态系统中蓬勃发展所必需的。除了一般的管家功能外,最小的基因组还包含许多功能未知的基因,这些基因在测序的基因组中很少见,可能在肠道中特别需要。
除了提供人类肠道微生物组的全球视野之外,我们已经建立的广泛的基因目录使微生物基因与人类表型乃至未来更广泛的人类生活习惯之间的关联研究成为可能,并考虑了环境,包括饮食、生到老年。我们期望这些研究将使人们对人类生物学的了解比我们目前所拥有的更加全面。
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA