NAR:rrnDB-16S拷贝数校正数据库
陕西省微生物研究所 常帆
主要研究方向为土壤微生态,同时负责服务器维护和相关流程搭建。
写在前面
看到一篇文献:Microbial functional trait of rRNA operon copy numbers increases with organic levels in anaerobic
digesters(ISME:厌氧消化器中rRNA操纵子拷贝数的微生物功能特性)(DOI https://doi.org/10.1038/ismej.2017.135),文中提到利用rrnDB数据库通过拷贝数矫正微生物组相对丰度。就想学习一下rrnDB数据库。
引文如下,目前Google统计引用239次。
Stoddard S.F, Smith B.J., Hein R., Roller B.R.K. and Schmidt T.M. (2015) rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Research 2014; doi: 10.1093/nar/gku1201 [PMID:25414355]
rrnDB数据库简介
rrnDB数据库,全称ribosomal RNA operons (rrn) DataBase,是一个收集了NCBI全基因组数据的细菌和古菌的16S拷贝数的数据库。可以通过NCBI或RDP数据库的ID进行检索(Search)和评估(Estimate)。16S基因在一个物种中会有多拷贝以维持生存安全,在进行高通量测序时,因为拷贝数基数不同,PCR后测序会放大这种基数效应,造成物种的reads数产生偏差。rrnDB数据库就是基于物种全基因组的拷贝数进行评估和矫正的一款在线数据库和工具。
数据库网址:https://rrndb.umms.med.umich.edu/
截止2018年11月24日,数据库最后一次更新是2018.9.25,说明作者在一直维护。
图1. 网站主页
网站工具简介
网站有详细的说明文档,在Manual中。其中主要的两个功能是Search和Estimate。
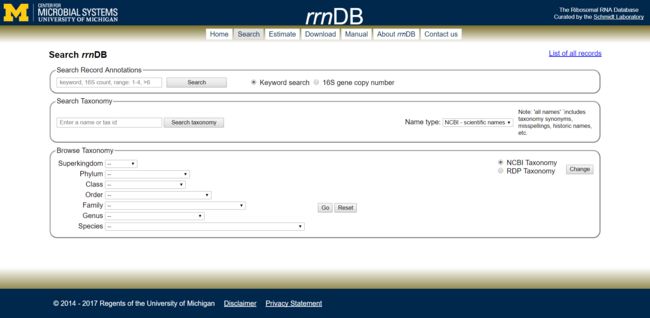
Search:拷贝数查询
图2. Search工具
- Search:用于搜索某个或者某些自身研究中感兴趣的菌株的16S rRNA基因拷贝数,搜索栏分3部分:
- Search Record Annotations:总搜索栏,可以输入不同分类水平菌株keyword直接搜索对应库中该菌的16S rRNA拷贝数。也可以按照拷贝数(切换选项至:16S gene copy number)来搜索数据库。搜索规则很简单:例如需要查看数据库中有6拷贝数的菌株,就输入数字6;需要查看3-5个拷贝数或大于5个拷贝数的菌,则直接输入3-5或者>6,搜索完毕会给出数据库中详细信息和简单统计:
- Search Taxonomy:可以通过菌株在NCBI和RDP中的名称进行搜索。
- Browse Taxonomy:是浏览器形式的搜索方式,可以通过不同分类水平进行搜索。
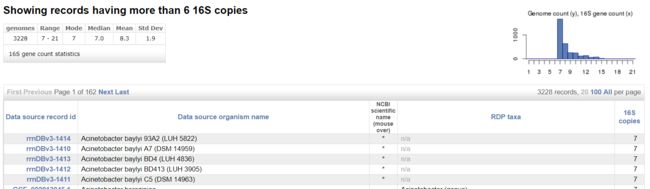
图3. Search Record Annotations搜索 >6 的结果
可以看到搜索了3228个NCBI上传的基因组信息,大于6个拷贝数分布从7-21,中位数7,平均值8.3等信息,右边还给出了简单的图形。说明 > 6个16S rRNA基因拷贝数的菌大部分为7个拷贝数。下面就是各菌的详细信息:id,菌株名称,NCBI名称,RDP注释和具体拷贝数:其中n/a表示找到23S rRNA但是没有16S的记录。
Estimate:序列物种注释和拷贝数校正
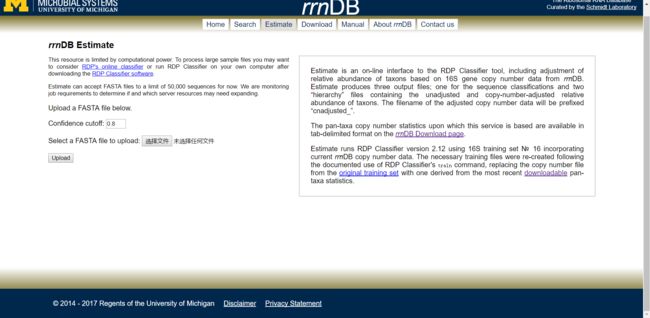
图4. rrnDB Estimate页面
- Estimate:是在线的RDP分类器工具,通过rrnDB的16S基因拷贝数数据矫正和估计OTU的相对丰度。在线工具支持上传最多5万条OTU序列的fasta格式文件,再多可以下载相关数据库在本地服务器中使用。工具产生三个输出文件:
- Classification file:(otus.tsv)用于序列分类。
- Hierarchical abundance file:(otus.hier.tsv):包含未调整的OTU的相对丰度。
- Copy-number adjusted hierarchichal abundance file(cnadjusted_otus.hier.tsv):调整后的拷贝数数据,文件名以cnadjusted_为前缀。
数据库可以设置cut_off value:默认0.8。然后选择文件,点击upload上传数据。
程序开始进行评估(estimate),等待结果。结果生成上面提到的3个文件(图5),且3个文件会保留3个月,将连接复制下来可在任何地方再次下载该数据。
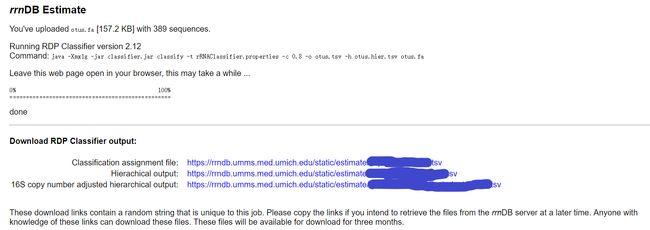
图5. rrnDB Estimate结果
结果文件说明
otus.tsv:就是对OTU文件进行RDP分类注释。对OTU按照“界门纲目科属种”进行注释,并有每一层注释的置信值。要注意这里会注释到“sub”分类水平(标记的一行),导致表格不整齐,后续使用需注意。
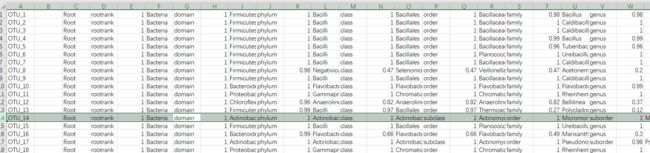
图6. otus.tsv
otus.hier.tsv和cnadjusted_otus.hier.tsv:未矫正和矫正过的16S rRNA拷贝数数据表格,表格形式完全一样,所以放在一个文档里对应展示:数据是按照相对丰度和分类水平进行排序,本例中首先统计厚壁菌门Firmicutes下的不同分类水平的相对丰度,然后是拟杆菌门Bacteroidetes。
矫正策略是利用表格2 otus.hier.tsv(左侧数据)的不同分类水平下各自的平均拷贝数对序列进行矫正生成表格3 cnadjusted_otus.hier.tsv(右侧数据),具体方法大家可以参考作者论文 https://www.ncbi.nlm.nih.gov/pubmed/25414355。
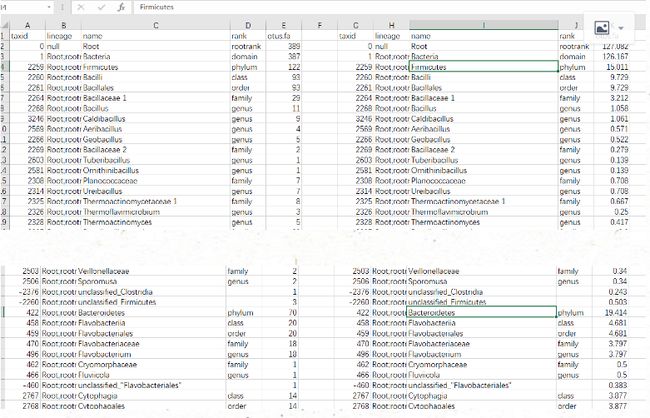
图7. 矫正前、后的物种-丰度表
得到了不同分类水平下矫正过的相对丰度,就可以对表格进行整理,统计不同分类水平下的相关数据了。
文献的使用实例
那么回到刚才提到的ISME 文献中,它究竟怎么使用相关数据的呢?文中提到“We calculated the abundance-weighted average rRNA operon copy number of OTUs for each sample (Figure 2a).”应该是利用OTU注释到的分类信息搜索rrnDB数据库,得到每一个OTU的16S rRNA拷贝数,再利用丰度信息进行丰度信息和拷贝数的加权平均得到的最终样品拷贝数的加权平均数。
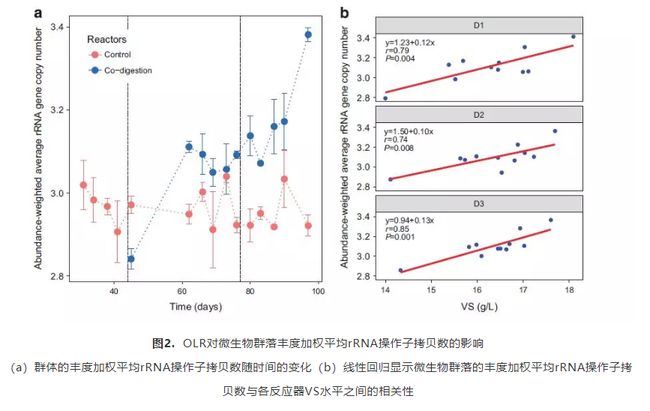
图8. rRNA加权平均值与发酵时间、挥发性固形物(VS level)展示
从浏览器search不同分类下的拷贝数太慢了,可以先下载网站最新的数据库:
rrnDB-5.5_pantaxa_stats_RDP.tsv.zip(在download中,截至2018.11.9),数据库文件格式如下:
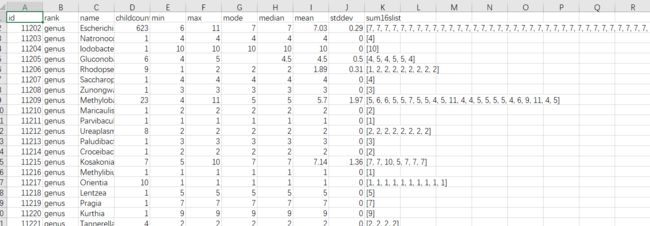
图9. 数据库文件
rank:分类水平,name:注释名称,childcount:所有该分类下共有多少个拷贝数,后面就是最大最小中位数平均值方差,最后是sum16slist拷贝数列表。
所以文章实现方法应该是:将注释表格1和矫正丰度表格3和数据库表格进行属水平注释的合并,然后抽出OTU行、注释行、矫正过的相对丰度行、16S拷贝数的均值行,根据相对丰度和均值行进行加权平均(即算术平均值的加权平均值),得到了最终样本的OTU水平拷贝数的加权平均值。再根据时间或者VS level进行作图。查看了文章的附件“In brief, for each OTU, the mean operon copy number (if available) of the immediate child taxa was used as the mean copy number.” 确实是以拷贝数的均值行作为注释OTU的16S拷贝数。
同时网站和工具还可以进行物种注释、依赖拷贝数矫正相对丰度,具体的用法大家可以多阅读相关参考文献多思考。
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2400+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA