Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid
脑脊液中病原体检测的临床宏基因组测序测定法的实验室验证
简述:
1.开发了一种经过验证的临床mNGS检测方法,用于在许可的微生物实验室中诊断脑脊髓液(CSF)引起的脑膜炎和脑炎的感染原因。
2.开发了定制的生物信息学管道SURPI+,以快速分析mNGS数据,生成检测到的病原体的自动摘要,并提供用于评估和解释结果的图形用户界面。
3.mNGS提供了一种全面的方法,通过该方法可以在一次测定中准确鉴定几乎所有潜在病原体-病毒,细菌,真菌和寄生虫。
4.将mNGS测试迁移到临床微生物学实验室仍然面临许多挑战。
(1)缺乏用于mNGS临床验证的既定蓝图
(2)难以将病原体从殖民者或污染物中区分开
(3)缺乏专为临床诊断使用的生物信息学软件
(4)注重质量和全面性的可获得的参考数据库
(5)临床实验室改进修正案(CLIA)环境中,患者诊断测试固有的法规遵从性要求
mNGS分析工作流程
A:核酸提取;B:文库生成,PCR扩增,上机测序;C:生物信息分析流程 SURPI+
补充描述:
A、敲击珠子、添加内部对照来提取CSF以回收病毒,细菌,真菌和寄生虫核酸。并分成两等份,以构建独立的DNA和RNA文库。通过基于抗体的甲基化宿主DNA去除(对于DNA文库)或DNase处理(对于RNA文库)。
B、基于转座子的文库构建,PCR扩增微生物序列得以富集,文库定量外部,加入阳性控制(PC)和阴性非模式控制(NTC)对照。上机测序,每个文库至少500万个reads。阴性对照用于监测外部或试剂微生物污染和交叉样品污染。阳性对照可用于检测核酸提取,文库制备过程或信息学方面的性能故障。
C、通过修饰adapter和去除低质量/低复杂性序列,对reads进行预处理,去除人类宿主reads,然后将剩余的微生物reads按类别,科或种分类。对于病毒,将reads映射到最接近的匹配基因组,以识别非重叠区域。对于细菌,真菌和寄生虫,将计算百万分之一(RPM)比率(RPM-r)度量,为了辅助分析,将生成自动结果摘要,原始/标准化读取计数的热图以及覆盖率/成对身份图,以用于检查和临床解释。
SURPI+软件可视化
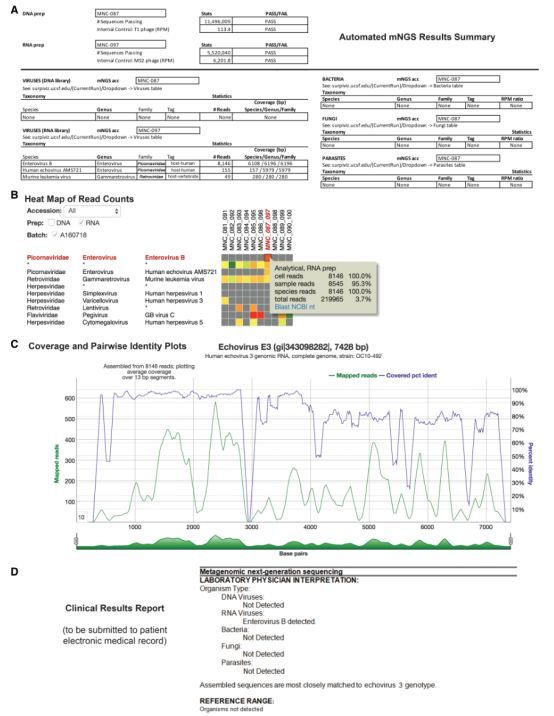
SURPI+提供了有助于临床解释和图形可视化的工具(SURPIviz软件包)
(A)mNGS结果摘要,提供QC运行指标和整体临床解释
(B)与检测到的病原体相对应的比对reads的热图
(C)针对NCBI GenBank核苷酸(nt)数据库中最接近匹配的微生物参考基因组或序列的比对命中的覆盖图
(D)实验室医师准备的临床结果报告
筛选病原体的阈值
对于病毒,阈值标准基于对不重叠reads的检测≥3个不同基因组区域。
细菌,真菌和寄生虫 RPM-r = RPMsample/RPMNTC
(RPM-r≥10)(下图验证阈值为10)

mNGS分析的性能特征

概述
1.检测限(LOD)(定义为检测到95%的阳性样品的最低浓度)
2.精度:测定间的可重复性(20),测定内的可重复性(3)
3.准确性:95例CSF患者样本(常规临床检测发现73例阳性,检测出79种生物体,22例阴性。)被用作mNGS分析。
4.干扰:评估了人类DNA和RNA,红细胞溶血以及同一属相关物种的混合物对mNGS分析性能的影响
5.稳定性:分别在4°C保持0、2、5和6 d并经过三个冻融循环的PC的重复分析表明,可以检测到所有生物
6.挑战研究:20例CSF样本,对mNGS分析进行盲法评估
mNGS相对于CSF临床测试的准确性
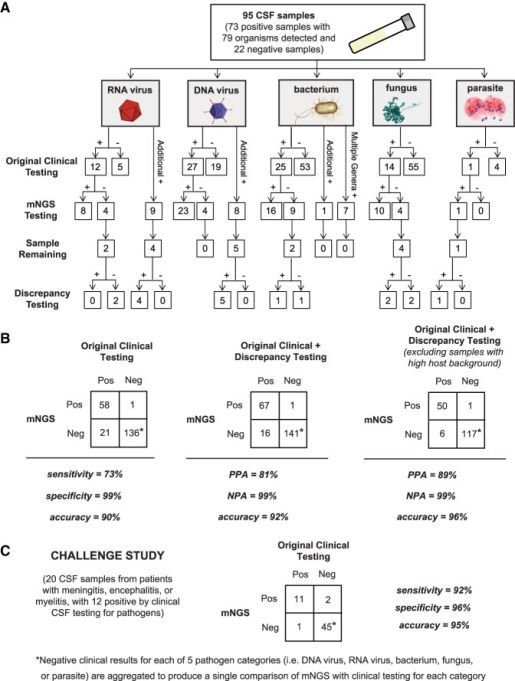
(A)在准确性研究中评估的样品结果流程图。结果按生物类别(RNA病毒,DNA病毒,细菌,真菌和寄生虫)分开。显示的是通过临床检测呈阳性或阴性的样品数量(第一行),mNGS结果与阳性临床结果之间的一致性以及mNGS产生的其他阳性检测物(第二行),以及在具有足够残留量的样品中(第三行)正交确认测试的结果(第四行)。
(B)2×2列联表比较了mNGS相对于CSF临床测试的性能。所使用的综合参考标准是原始临床测试(左),原始临床和差异测试的组合(中),排除高宿主背景样本后的原始临床和差异测试的组合(右)。(PPA)阳性预测一致性,(NPA)阴性预测一致性。
(C)2×2列联表显示了挑战研究的结果。通过mNGS分析了20个CSF样本,并与常规临床测试结果进行了比较。
ps:
灵敏度=真阳性人数 /(真阳性人数+假阴性人数)*100%。
特异度=真阴性人数 /(真阴性人数+假阳性人数)*100%。
准确度=(真阳性人数+真阴性人数)/(真阳性人数+假阳性人数+真阴性人数+假阴性人数)*100%
为验证准确性检查临床数据
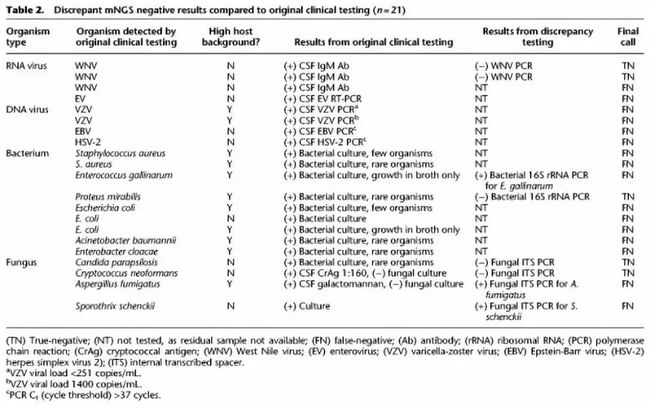
最初将21例病例归为mNGS假阴性; 21个中的8个具有足够的残留CSF量可用于差异测试。在这8例mNGS阴性病例中,有5例(2例WNV,变形杆菌,假丝酵母和新型隐球菌)为阴性,因此经差异检测后重新分类为真阴性。通过后续PCR检测,八分之三的mNGS阴性病例为阳性,因此认为是真正的mNGS假阴性结果。
准确性图的B部分中,左->中的21->16少的5例阴性

在18例病例中,mNGS检测到了其他未经临床检验的生物(表3)。9例(4例HIV,1例CMV,2例EBV,1例HSV-1、1例HHV-6)有足够的CSF样本可用于差异检测,后续的PCR检测证实了这9例病例中mNGS阳性。被重新分类为真阳性病例
准确性图的B部分中,左->中的58->67多的9例阳性
总结延伸
影响mNGS临床敏感性的因素包括:
(1)仅通过血清学诊断的病例(例如WNV),
(2)使用临床检测作为不完善的“金标准”,有些样品可能代表对污染的假阳性检测(例如,粪肠球菌仅从肉汤中生长)
(3)分析残留的生物库临床样品以进行mNGS准确性测试,以及先前冻融步骤的降解可能会降低灵敏度
(4)使用可靠的预先建立的阈值以最大程度地减少假性阳性检测
(5)人类宿主背景(例如,高CSF胞吞作用)在限制敏感性中的作用
mNGS数据的二次分析结果包括
(1)用于预测抗生素或抗病毒抗药性突变预测的基因组装配
(2)基因型和菌株水平鉴定的系统发育分析
(3)披露低于正式报告阈值的潜在病原体reads
因此,mNGS发现的临床相关性可以有效地传达给医生,从而可能为患者的管理和治疗提供后续信息,也可能为公共卫生监测和疾病暴发调查提供信息。
开发的工作流软件SURPI+
下载地址:https://github.com/chiulab/SURPI-plus-dist
包含软件如下:
i.NCBI Blast suite v2.7.1Link: http://blast.ncbi.nlm.nih.gov/Blast.cgi
ii.Bowtie2 v2.3.2Link:http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
iii.cutadapt v.1.9.1-1build1Link: https://cutadapt.readthedocs.io/en/stable/
iv.fastQValidator v1.0.14Link: https://github.com/statgen/fastQValidator
v.gt v.1.5.9Link: http://genometools.org
vi.PRINSEQ-lite v0.20.4Link: http://prinseq.sourceforge.net
vii.seqtk v1.3Link: https://github.com/lh3/seqtk
viii.SNAP v1.0dev100Link: http://snap.cs.berkeley.edu/
ix.vmtouch v1.3.1Link: https://github.com/hoytech/vmtouch