1、RRBS简介
RRBS即reduced representation bisulfite sequencing:
第一步:使用MSPI内切酶处理,其识别序列是CCGG,切完之后,双链序列如下:
CGG--------------------------------C
C--------------------------------GGC
第二步:自动补齐
CGG--------------------------------CCG
GCC--------------------------------GGC
第三步:bisulfite conversion
将双链中没有甲基化的C变为U,甲基化的C保持不变,这样转化之后,原来互补的两条双链就不互补了
2、数据预处理
2.1 fastqc质量检测
fastqc -t 2 -o ./00_fastqc R1.fastq R2.fastq
主要看Q30的百分比,以及reads中有无接头
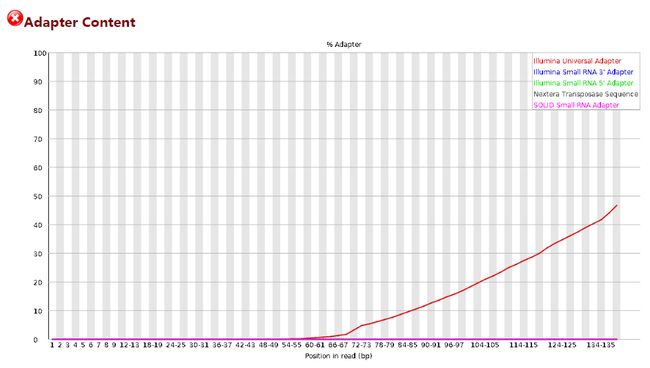
从图中可看出,reads中只有illumina universal adapter,因此在后续的处理中只需处理这一种接头
2.2 trim_galore去除接头
trim_galore --phred33 --illumina --stringency 3 -e 0.1 \
--gzip --length 35 --rrbs --fastqc -o ./cut_adapter \
--paired R1.fastq R2.fastq
详细参数使用trim_galore -h查看,这里特别解释一下--rrbs这个参数,是专门用于用MspI处理的RRBS数据,加上这个参数,在切除接头之后会自动切除2bp的碱基,即上面说的自动补齐的那两个碱基。另外--illumina这个参数是用来切除illumina universal adapter(AGATCGGAAGAGC)的,另外参数--nextera切除nextera adapter(CTGTCTCTTATA),--small_rna切除Illumina Small RNA 3' Adapter(GATCGTCGGACT)
3、使用bismark转换基因组序列
参考基因组下载:
https://support.illumina.com/sequencing/sequencing_software/igenome.html
bismark_genome_preparation --path_to_bowtie dir_bowtie --genomic_composition \
--verbose dir_ref dir_hg38.fa
4、序列比对
bismark dir_ref -1 $R1 -2 $R2 --path_to_bowtie $bowtie \
-o ./01_align --rg_tag --rg_id $fname --rg_sample $fname \
--unmapped --prefix $fname --basename $fname --nucleotide_coverage
比对好的结果为bam格式的,其内容与BWA比对后的bam文件稍微有些不同
上面是一条bam文件的记录,共17列,下面分别解释一下各列的含义:
第1列:
E00500:282:HN3M3CCXY:7:1101:8613:2628_1:N:0:ATCACG
#reads编号,_1代表来自原来的R1,后面的ATCACG为index序列第2列:
此列为一数值,具体解释如下图
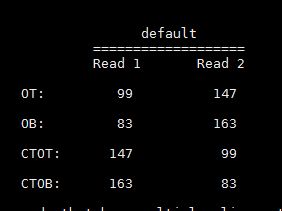
相同数值对于R1和R2来说代表不同的链,此外,这些数值也是由多个值相加所得,下面这几个标签都是2的n次方,随机挑选其中的几个,他们的和是唯一的:
| 1 | 代表这个序列采用的是PE双端测序 |
| 2 | 代表这个序列和参考序列完全匹配,没有插入缺失 |
| 4 | 代表这个序列没有mapping到参考序列上 |
| 8 | 代表这条序列的另一端序列没有比对到参考序列上,如果这条是R1,它对应的R2端没有比对上 |
| 16 | 代表这个序列比对到参考序列的负链上 |
| 32 | 代表这个序列对应的另一端序列比对到参考序列的负链上 |
| 64 | 代表这个序列是R1端序列 |
| 128 | 代表这个序列是R2端序列 |
| 256 | 代表这个序列不是主要的比对,一条序列可能比对到了多个位置,只有一个是首要的比对位置 |
| 512 | 代表这个序列在QC时失败了,被过滤不掉了 |
| 1024 | 代表这个序列是PCR重复序列 |
| 2048 | 代表这个序列是补充的比对,不常用 |
第3-4列:
chr4 184651014
代表这条reads比对到4号染色体的184651014这个位置
第5列:
为一数值,Mapping quality,比对的质量分数,越高说明该read比对到参考基因组上的位置越唯一
第6列:
CIGAR值,碱基匹配上的碱基数。
| M | match/mismatch | I | insert | |
| D | deletion | N | skipped(跳过这段区域) | |
| S | soft clipping(被剪切的序列存在于序列中) | H | hard clipping(被剪切的序列不存在于序列中) | |
| P | padding(填充) |
135M
代表135个碱基在比对时完全比对
3S6M1P1I4M
代表前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的
第7列:
MRNM(chr),mate的reference sequence name,实际上就是mate比对到的染色体号,若是没有mate,则是*,若为=则代表与此条比对到相同的染色体上。
第8列:
mate position,mate比对到参考序列上的第一个碱基位置,若无mate,则为0
第9列:
insert size
第10列:
Sequence,就是read的碱基序列,如果是比对到负链上则对read进行了reverse completed
第11列:
ASCII,read质量的ASCII编码
第12列:
NM-tag,与bowtie中NM:i相同。read string转换成reference string需要的最少核苷酸的edits:插入/缺失/替换
第13列:
XX-tag,对错配的描述,不包括indel(插入和缺失)
XX:Z:2C4C2C6C1C1AC18C3C15C4C2CC14
表示2个碱基完全匹配,一个C替换,接着4个碱基完全匹配,一个C发生替换
第14列:
XM-tag,methylation call string
XM:Z:........xz.z....hx............x....h.hh....xz....xz...x....h.hh..hhhx....z...h......h

CHG、CHH、CG代表甲基化的三种模式
CG:C后面接的是G
CHG:C后面跟了任意一个碱基,再接上一个G
CHH:C后面跟的是除了G以外的任何碱基
第15列:
XR-tag,read conversion state for the alignment 。
共两种转换:CT和GA,GA就是指将read里的所有G转换成A
第16列:
XG-tag,genome conversion state for the alignment。
共两种:GA和CT。CT是指将全基因组上所有的C转换成T
第17列:
表示样本名称,是在做比对时通过--rg_tag自己设置的
5、去除重复序列
deduplicate_bismark --paired --bam bam_file --output_dir dir_bamfile
注:RRBS其实是不需要做这一步的
6、抽提出甲基化的统计信息
bismark_methylation_extractor --paired-end --report --output dir --gzip --bedGraph bam_file
产生的结果文件如下:
CHG_OB_FF01N_ATCACG_S9_L007_pe.deduplicated.txt.gz
CHG_OT_FF01N_ATCACG_S9_L007_pe.deduplicated.txt.gz
CHH_OB_FF01N_ATCACG_S9_L007_pe.deduplicated.txt.gz
CHH_OT_FF01N_ATCACG_S9_L007_pe.deduplicated.txt.gz
CpG_OB_FF01N_ATCACG_S9_L007_pe.deduplicated.txt.gz
CpG_OT_FF01N_ATCACG_S9_L007_pe.deduplicated.txt.gz
FF01N_ATCACG_S9_L007_pe.deduplicated.bedGraph.gz
FF01N_ATCACG_S9_L007_pe.deduplicated.bismark.cov.gz
FF01N_ATCACG_S9_L007_pe.deduplicated.M-bias.txt
FF01N_ATCACG_S9_L007_pe.deduplicated_splitting_report.txt