在前面盲人摸象--single cell sequencing的Seurat学习--详细版这篇文章之后,我膨胀了!感觉好想好想学习,因此,依据之前阅读单细胞文献的经历,将自己感兴趣的教程打算过一遍:cell-type specific responses,即不同组别的细胞放在一起比较。这个方法的应用是非常广泛的,尤其是肿瘤组织和癌旁组织的重叠比较。
官网教程地址:Tutorial: Integrating stimulated vs. control PBMC datasets to learn cell-type specific responses
1. 环境准备+数据载入
准备Seurat,SeuratData,cowplot这三个包,需要介绍的SeuratData是一个Seurat团队专门整理的一个数据库,可用于training,详情可点击SeuratData
1.1 准备环境
# 缺啥就安装啥,SeuratData的安装方法在GitHub上看到
devtools::install_github('satijalab/seurat-data')
library(Seurat)
library(SeuratData)
library(cowplot)
1.2 载入数据
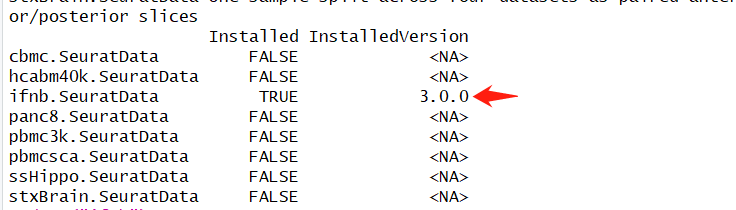
InstallData("ifnb")
# 使用AvailableData()查看目前SeuratData可用的data
AvailableData()
# 可以看到只有ifnb.SeuratData这一项是TRUE,因为我刚刚下载了
# ifnb.SeuratData 是IFNB-Stimulated and Control PBMCs
1.3 数据预处理
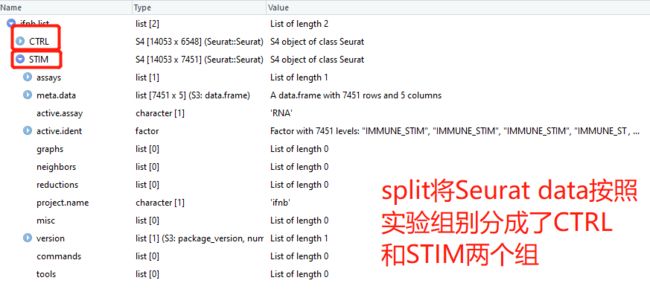
按照实验组别拆分数据
data("ifnb") #231.9 Mb
class(ifnb)
# [1] "Seurat"
# attr(,"package")
# [1] "Seurat"
ifnb.list <- SplitObject(ifnb, split.by = "stim")
#按照试验分组split数据,在ifnb数据中可看到stim其实是实验分组
# stim的意思应该是stimulation
一键操作完成normalization和差异分析
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst",
nfeatures = 2000)
})
# 秒啊
# 可见别人代码的优美
2. Perform integration 执行集合
使用findintegrationanchor函数来识别锚点,该函数以Seurat对象的列表作为输入,并使用这些锚点将两个数据集以IntegrateData集成在一起。但为什么要拆开再合并?大约是每个样本都要单独进行normalization和差异分析吧,完成之后再合并,依照集合时选出的2000个anchors进行筛选,用于下一步的操作。
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list,
dims = 1:20) # 选用20个PC
# 这里说的anchors是什么呢?
 image.png
image.png
解答anchors是什么
immune.combined <- IntegrateData(anchorset = immune.anchors,
dims = 1:20)
# 木得感情的抄代码
# 如果纠结于为什么是这个代码,或者为什么使用这个参数将会阻碍学习的进度
# 木得感情的使用代码
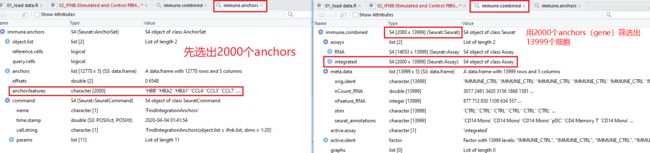 image.png
image.png
(1)2000个gene筛出13999个细胞,如果是普通的矩阵,那应该是行名是gene ID,列名是barcodes(每个barcode代表一个细胞),就应该是2000x13999这样的大矩阵。
(2)除了这样的信息之外,13999个细胞,每一个都会有自己的分组,是CTRL(control)还是STIM(stimulation)组,所以应该还会有一个,13999行x分组列的矩阵或者data.frame。
(3)但是好似这边没有提到线粒体基因的比例问题,这部分应该是在非常早期的时候,做QC筛选已经normalize过了,所以这边没有提到吧。
3. Perform an integrated analysis
准备数据,设置默认的assay。由于immune.combined有RNA和integrated两个assay,需要设置默认为integrated。
DefaultAssay(immune.combined) <- "integrated"
Seurat::Assays(immune.combined)
# immune.combined里面有RNA和integrated两个assay
# 将immune.combined的默认assay设置为"integrated"
常规的可视化和聚类流程:先缩放,再确认PC数,确认数据维度,可视化绘图
# Run the standard workflow for visualization and clustering
# 数据可视化和聚类的标准流程
# 缩放数据
immune.combined <- ScaleData(immune.combined, verbose = FALSE)
# PCA分析
immune.combined <- RunPCA(immune.combined, npcs = 30, verbose = FALSE)
# t-SNE and Clustering
immune.combined <- RunUMAP(immune.combined, reduction = "pca", dims = 1:20)
# 它选了20个PC
# 输入到下一步骤
immune.combined <- FindNeighbors(immune.combined, reduction = "pca", dims = 1:20)
# 输入到下一步骤
immune.combined <- FindClusters(immune.combined, resolution = 0.5)
## 可视化,分别画图再合并
p1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p2 <- DimPlot(immune.combined, reduction = "umap", label = TRUE)
plot_grid(p1, p2)
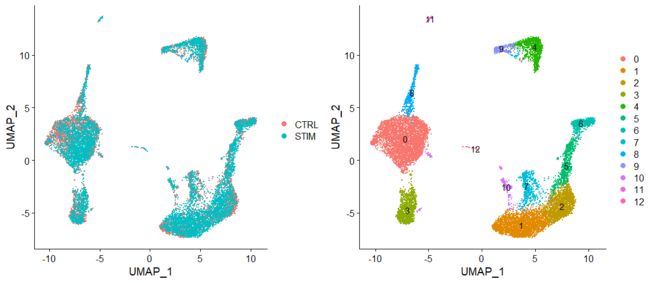 image.png
image.png
左边的图是按照分组显示颜色;右边图是按照cluster数目label,但是cluster数目是怎么计算出来的呢?在这里我盲猜是因为resolution的原因,前面设置了resolution=0.5,我把resolution改为0.1,看看会如何:
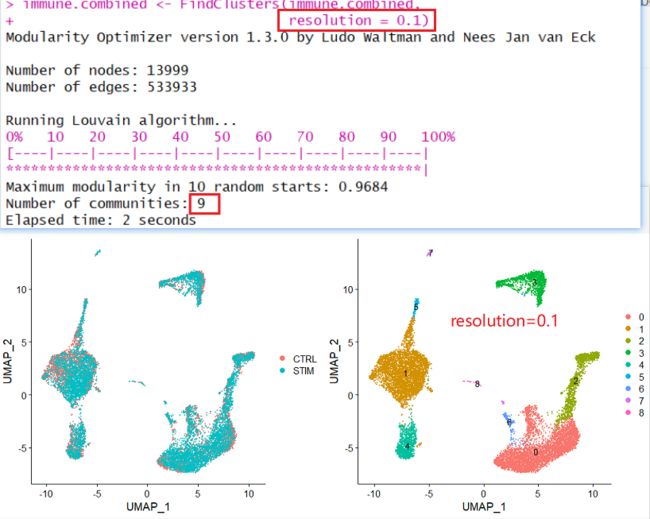 image.png
image.png
除了将两组合并在一起显示之外,还可以将CTRL和STIM分来绘图
DimPlot(immune.combined, reduction = "umap", split.by = "stim")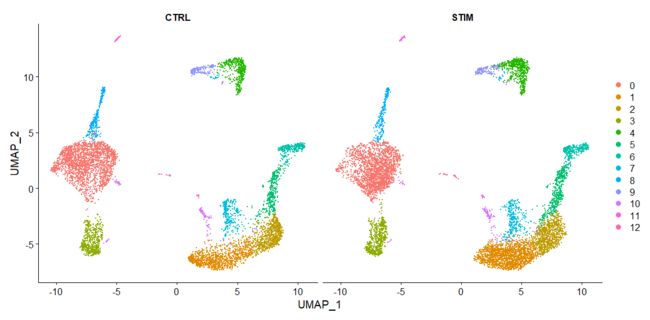 image.png
image.png
点评:来到这里基本上已经完成数据前期处理了,由于数据是Seurat整理好的,QC,过滤,线粒体基因等处理已经没有了,直接开始就是导入数据,拆分normalization和寻找高差异基因。拆分再合并的好处是什么?可以降低组间差异吧。如果直接放在一起又该如何?
4. Identify conserved cell type markers
使用findconservedmarker函数识别在不同条件下cluster的标记基因,需要注意这是两组的集合,因此原先提到过的FindAllMarkers和FindMarkers就不适用了。findconservedmarker函数对每个数据集/组执行差异基因表达测试,并使用来自MetaDE R包的meta analysis方法组合p值。例如,我们可以计算出在cluster 7 (NK细胞)中,无论刺激条件如何,都是保守标记的基因。即:需要找到不受实验刺激影响的conserved(保守的)基因,这样的基因才能作为cluster的marker。
DefaultAssay(immune.combined) <- "RNA"
# 提前知道cluster是cluster 7咯
nk.markers <- FindConservedMarkers(immune.combined,
ident.1 = 7, grouping.var = "stim", verbose = FALSE)
head(nk.markers)
# 可视化前几个nk.markers
FeaturePlot(immune.combined,
features = c("SNHG12", "HSPH1","NR4A2", "SRSF2", "BATF", "CD69"),
min.cutoff = "q9")
# 小提琴图
VlnPlot(immune.combined,
features = c("SNHG12", "HSPH1","NR4A2", "SRSF2", "BATF", "CD69"),
ncol = 3)
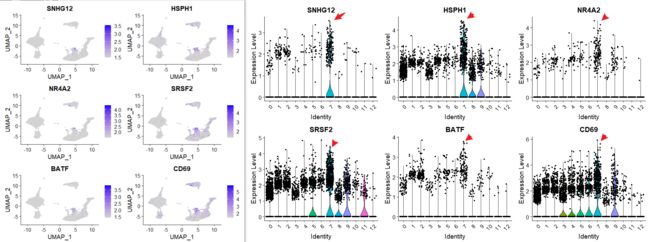

注意:查看每个基因在不同cluster中的贡献情况,使用函数DotPlot,画面非常舒适
# Doplot ------------------------------------------------------------------
Idents(immune.combined) <- factor(Idents(immune.combined),
levels = c("Mono/Mk Doublets", "pDC",
"Eryth", "Mk", "DC",
"CD14 Mono", "CD16 Mono",
"B Activated", "B", "CD8 T",
"NK", "T activated",
"CD4 Naive T", "CD4 Memory T"))
# level是13个cluster的名字
markers.to.plot <- c("CD3D", "CREM", "HSPH1", "SELL", "GIMAP5",
"CACYBP", "GNLY", "NKG7", "CCL5", "CD8A",
"MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A",
"VMO1", "CCL2", "S100A9", "HLA-DQA1", "GPR183",
"PPBP", "GNG11", "HBA2", "HBB", "TSPAN13", "IL3RA", "IGJ")
# 每个cluster选了2-3个高表达基因用于绘图
DotPlot(immune.combined, features = rev(markers.to.plot), cols = c("blue", "red"),
dot.scale = 8, split.by = "stim") + RotatedAxis()
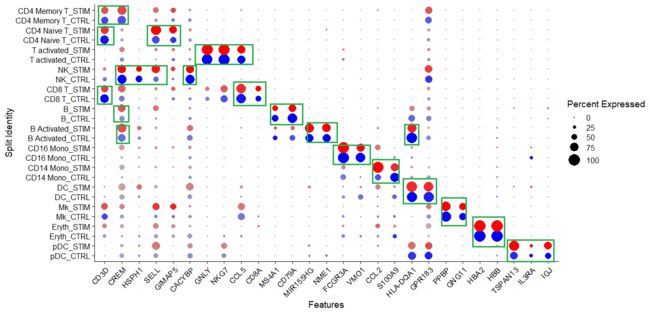
可以看到不同cluster的marker gene会有一定的重复
5. Identify differential expressed genes across conditions 鉴定由于实验引起的差异基因--
将IFNB刺激的细胞和control的细胞排列好后,可以开始进行对比分析,看看IFNB刺激后的不同之处。一种广泛观察这些变化的方法是绘制IFNB刺激细胞和control细胞的平均表达,并在散点图上寻找视觉异常的基因。在这里,我们取IFNB刺激和control的naive T细胞和CD14单核细胞群体的平均表达,并生成散点图,突出显示对IFNB刺激有显著反应的基因。
library(ggplot2)
library(cowplot)
theme_set(theme_cowplot()) # 主题是cowplot
t.cells <- subset(immune.combined, idents = "CD4 Naive T")
Idents(t.cells) <- "stim"
avg.t.cells <- log1p(AverageExpression(t.cells, verbose = FALSE)$RNA) # 平均
avg.t.cells$gene <- rownames(avg.t.cells)
cd14.mono <- subset(immune.combined, idents = "CD14 Mono")
Idents(cd14.mono) <- "stim"
avg.cd14.mono <- log1p(AverageExpression(cd14.mono, verbose = FALSE)$RNA)
avg.cd14.mono$gene <- rownames(avg.cd14.mono)
## 寻找STIM相对于CTRL异常升高的gene
avg.t.cells[["fold"]] <- avg.t.cells$STIM/avg.t.cells$CTRL
genes.to.label = c("ISG15", "LY6E", "IFI6", "ISG20", "MX1",
"IFIT2", "IFIT1", "CXCL10", "CCL8") # 从哪里知道的?
p1 <- ggplot(avg.t.cells, aes(CTRL, STIM)) + geom_point() + ggtitle("CD4 Naive T Cells")
p1 <- LabelPoints(plot = p1, points = genes.to.label, repel = TRUE)
p2 <- ggplot(avg.cd14.mono, aes(CTRL, STIM)) + geom_point() + ggtitle("CD14 Monocytes")
p2 <- LabelPoints(plot = p2, points = genes.to.label, repel = TRUE)
plot_grid(p1, p2)
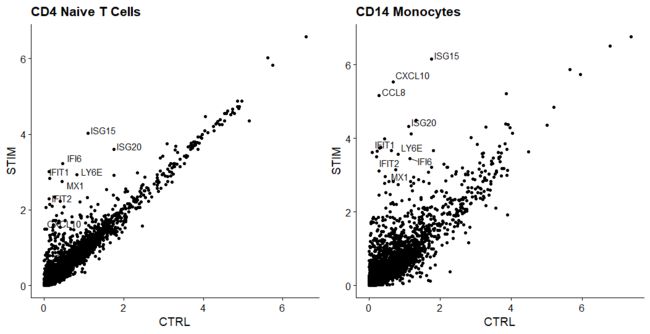
可见,许多相同的基因在这两种细胞类型中都被上调,可能代表了一种保守的干扰素反应途径。因为我们有信心在不同的条件下识别出共同的细胞类型,我们可以问相同类型的细胞在不同的条件下发生了什么基因变化。首先,我们在元数据中创建一个列来保存细胞类型和刺激信息,并将当前事件转换到该列。然后,我们使用findmarker找到受刺激细胞和对照组B细胞之间不同的基因。请注意,这里显示的许多顶部基因与我们之前绘制的核心干扰素响应基因相同。此外,我们看到的单核细胞和B细胞干扰素反应特异性的CXCL10等基因在这个列表中也显示出高度显著性。
immune.combined$celltype.stim <- paste(Idents(immune.combined),
immune.combined$stim, sep = "_")
immune.combined$celltype <- Idents(immune.combined)
Idents(immune.combined) <- "celltype.stim"
b.interferon.response <- FindMarkers(immune.combined, ident.1 = "B_STIM",
ident.2 = "B_CTRL", verbose = FALSE)
head(b.interferon.response, n = 15)
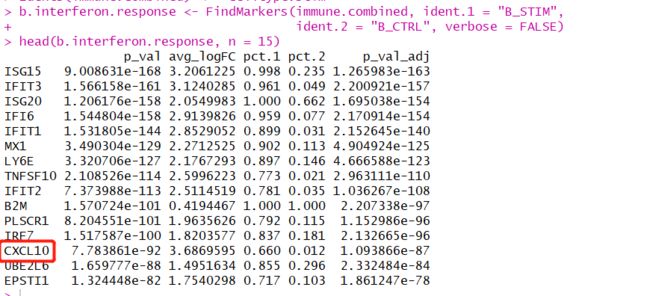 image.png
image.png
## 可视化
# CD3D,GNLY不受干扰素影响, IFI6 and ISG15是主要反应基因
FeaturePlot(immune.combined, features = c("CD3D", "IFI6"),
split.by = "stim", max.cutoff = 3, cols = c("grey", "red"))
# 不同的可视化方法
plots <- VlnPlot(immune.combined, features = c("LYZ", "ISG15", "CXCL10"),
split.by = "stim", group.by = "celltype",
pt.size = 0, combine = FALSE)
CombinePlots(plots = plots, ncol = 1)

结语
本教程的思路是如何将在不同条件下的样本放在一起分析,除了各自normalization和寻找差异基因之外。需要合并在一起做PCA和cluster,cluster之间可以有差异基因(但没有特别大的意义,主要是找到每个cluster的marker ),寻找cluster marker时要先找到保守的细胞marker(不受实验条件影响),再寻找由于实验处理引起的genes,这时候还是使用FindMarker,然后想各种办法去可视化。marker的类型有:高保守marker,cluster marker,细胞类型marker,由实验刺激引起的差异基因等等。理解思路,找到数据转换和处置的理由。