在最最最开始!放上借鉴引用的文章链接!(后边还有)
RNA-seq分析:从软件安装到富集分析详细过程
RNA-seq实战
第二次RNA-seq实战总结(2)-数据下载并进行数据处理
感谢各位大佬!
一、软件安装准备
1.原始数据下载软件Aspera
2.解压文件SRA-toolkit
3.比对软件hisat2
4.基因表达量软件htseq-count
(这是一个Python包,需要在Python2的环境下下载)
5.数据质量评价软件fastqc
6.数据处理过滤软件trimmomatics
7.samtools
以上软件均可用conda进行下载,只不过所需要的Python环境不太一样
- 首先在conda搜索软件
$ conda install +软件名
ps.conda可以一次性指定两个软件进行下载
pps.除了htseq-count需要在Python2的环境下下载之外,其余软件均可以在Python3的环境下载
关于conda,具体可参考conda管理生信软件一文就够
二、数据下载和处理
- 我选择的原始数据来源于
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50177
选择后四个samples,SUZ12和对照Ctrl,两个重复
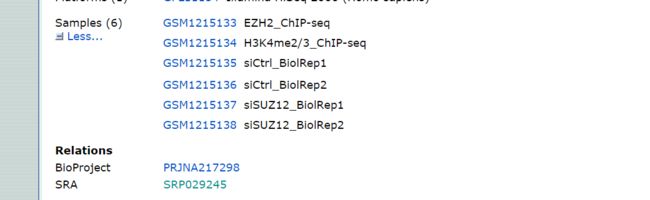
点击SRA Run Selector
我们需要的是红框里的四个数据

即需要的是SRR957677-SRR957680
1.下载sra数据
#我用prefetch来下载SRA文件,其实也可以用aspera connect来下载
#并且aspera connect更安全
#但是在下载过程中出现了一些问题,具体解决等我做完这次分析再去解决qaq
$ prefetch SRR957677 SRR957678 SRR957679 SRR957680
2.下载索引文件
- 下载hg19
$ mkdir reference & cd reference
$ cd reference
$ mkdir hg19
由于用wget下载很慢,显示要三天+
所以我再hisat2的官网复制下载链接在迅雷上下载的
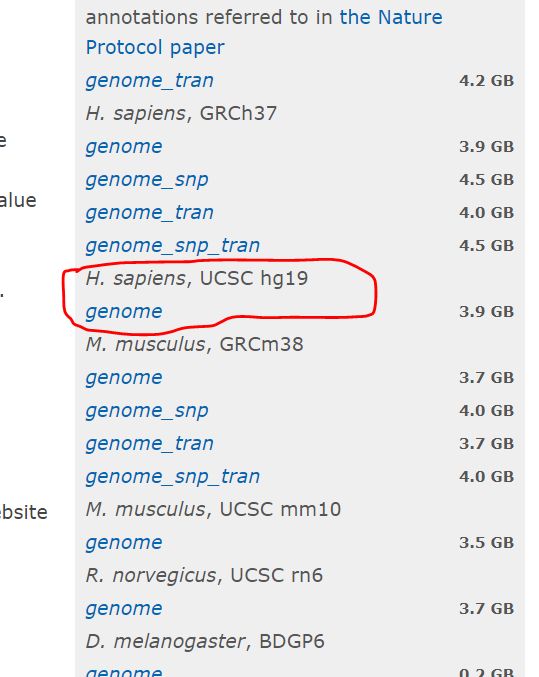
将下载好的文件传到hg19的文件夹下,然后解压
$ wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz & tar -zxvf chromFa.tar.gz
# # 将所有的染色体信息整合在一起,重定向写入hg19.fa文件,得到参考基因组
$ cat *.fa > hg19.fa
$ rm -rf chr*
ps:同样,这个我也是把文件下载下来再传输到我的Linux里的,因为文件也比较大。可以复制这个链接在浏览器打开,就会弹出下载框了;或者依然是在迅雷下载
pps:其实,解压了hg19之后,它自带了一个make_hg19.sh的文件,运行它来得到.fa文件也是可以的,不过还是那句话,wget下载速度太慢。。我还是先把chromFa.tar这个文件下载好了,再解压,再重定向
# 然后需要生成一个.fai的文件,需要用到samtools faidx
$ samtools faidx hg19.fa
- 下载基因组注释文件
$ wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_29/GRCh37_mapping/gencode.v29lift37.annotation.gtf.gz
$ mv gencode.v29lift37.annotation.gtf.gz hg19
$ unzip gencode.v29lift37.annotation.gtf.gz
三、转换成fastq文件,并进行质控以及过滤
1.fast-dump,转成fastq文件
$ mkdir fastq
$ fastq-dump --gzip --split-3 *.sra
$ mv *.fastq.gz ~/Seqs/experiment_report/fastq
2.fastqc质控
$ mkdir fastqc
#进到fastq文件夹
#-o后面接输出文件夹
$ fastqc *.fastq.gz -o ~/Seqs/experiment_report/fastqc
可以查看生成的HTML文件,来看测序质量如何。
例如,SRR957677
3.Trimmomatic过滤
#创建一个trim_out文件夹,存放过滤的文件
$ mkdir trim_out
#输出文件名字为SRR*****.fastq.clean.gz
$ java -jar ~/Biosoft/Trimmomatic-0.38/trimmomatic-0.38.jar SE -threads 2 -phred33 ~/Seqs/experimental_report/fastq/SRR957677.fastq.gz ~/Seqs/experimental_report/trim_out/SRR957677.fastq.clean.gz ILLUMINACLIP:/home/akuooo/Biosoft/Trimmomatic-0.38/adapters/TruSeq2-PE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20
这个过滤之后的文件如下图
其他三个文件同样如此,其实想设置一个for循环代码来处理,可能效率也更高。但是由于现在还是学术不精,还不太敢尝试,等我之后再多学一会再来试一试=-=
四、hisat2序列比对
for ((i=77;i<=80;i++));do hisat2 -t -x ./reference/hg19/genome -U ./trim_out/SRR9576${i}.fastq.clean.gz -S SRR9576${i}.sam ;done
这次参考了一下别人的代码,具体看第一篇引用的文章!
-x index : 参考基因组
双端测序:hisat2 -x hisat2_index -1 m1 -2 m2 -S name.sam
单端测序:hisat2 -x hisat2_index -U r1 -S name.sam
关于hisat2,可以看看RNA-Seq基因组比对工具HISAT2
ps:/hg19/genome的genome是个前缀!!不是文件名qaq我当时看的时候看错了,导致出现了无法用索引的情况
(((由于时间太久了……我终止了程序,准备明天早上再来搞……
大家如果准备比对的话,记得多留点时间……寝室断电了呜呜
-------------------------------------分割线--------------------------------------------
好的我继续了,由于昨天没弄完,今天只好重新开始qaq)))
9:02开始,看会持续多久(7点多起来弄,突然发现比对的文件用的不是过滤后的文件,哭了,又暂停了重新搞,大家一定一定要看清楚啊呜呜呜)
下面这个是运行的时候的代码,懒得截图了,上课前挂着等他比对,下课回来发现还有一个没有比对完……
Time loading forward index: 00:00:08
Time loading reference: 00:00:07
Multiseed full-index search: 01:15:04
20626594 reads; of these:
20626594 (100.00%) were unpaired; of these:
1244591 (6.03%) aligned 0 times
16906012 (81.96%) aligned exactly 1 time
2475991 (12.00%) aligned >1 times
93.97% overall alignment rate
Time searching: 01:15:26
Overall time: 01:15:34
Time loading forward index: 00:00:09
Time loading reference: 00:00:09
Multiseed full-index search: 00:32:49
8695598 reads; of these:
8695598 (100.00%) were unpaired; of these:
560868 (6.45%) aligned 0 times
7151059 (82.24%) aligned exactly 1 time
983671 (11.31%) aligned >1 times
93.55% overall alignment rate
Time searching: 00:33:11
Overall time: 00:33:20
Time loading forward index: 00:00:09
Time loading reference: 00:00:08
Multiseed full-index search: 01:04:44
19741163 reads; of these:
19741163 (100.00%) were unpaired; of these:
1303760 (6.60%) aligned 0 times
15831001 (80.19%) aligned exactly 1 time
2606402 (13.20%) aligned >1 times
93.40% overall alignment rate
Time searching: 01:05:06
Overall time: 01:05:15
Time loading forward index: 00:00:09
Time loading reference: 00:00:08
Multiseed full-index search: 01:04:44
19741163 reads; of these:
19741163 (100.00%) were unpaired; of these:
1303760 (6.60%) aligned 0 times
15831001 (80.19%) aligned exactly 1 time
2606402 (13.20%) aligned >1 times
93.40% overall alignment rate
Time searching: 01:05:06
Overall time: 01:05:15
Time loading forward index: 00:00:10
Time loading reference: 00:00:08
Multiseed full-index search: 01:37:13
24030038 reads; of these:
24030038 (100.00%) were unpaired; of these:
1407817 (5.86%) aligned 0 times
19747330 (82.18%) aligned exactly 1 time
2874891 (11.96%) aligned >1 times
94.14% overall alignment rate
Time searching: 01:37:34
Overall time: 01:37:44
可以看到,花了好久好久。。
五、htseq-count,reads计数
- 首先把Sam文件转成bam文件(这一步是参考了暴老师的RNAseq步骤)
$ mkdir bam
$ cd sam
# -@为设置线程数
$ for ((i=77;i<=80;i++));do samtools view -bt ./reference/hg19/hg19.fa.fai -@ 2 -o ../bam/SRR9576${i}.bam SRR9576${i}.sam 2>>samtools.log;done
- 然后对其进行排序(依旧参考暴老师)
#
$for ((i=77;i<=80;i++));do samtools sort -O bam -@ 2 -o SRR9576${i}.sort.bam -T tmp_samtools SRR9576${i}.bam 2>>samtools.log;done
这是我生成的文件
- 其实还可以建立索引,生成的文件可以用来进行
IGV可视化,我暂时先不弄继续下一步。
#生成索引文件
for ((i=77;i<=80;i++));do samtools index SRR9576${i}.sort.bam;done
-
htseq-count,计算比对到每个基因的短序列数目
$ mkdir counts & cd counts
$ for ((i=77;i<=80;i++));do htseq-count -r name -f bam ../bam/SRR9576${i}.sort.bam ../reference/hg19/gencode.v26lift37.annotation.gtf > ./SRR9576${i}.count;done
做到这一步,我发现我电脑磁盘空间只剩十几G了,记得做之前看一下自己的内存……别做到一半发现没内存了就麻烦了……其实可以把之前得到的那些文件删一些的,像什么fastq啊等等,但是我怕出错没敢删。
这一步大概花了接近两个小时
- 对结果进行统计
$ wc -l *.count
60497 SRR957677.count
60497 SRR957678.count
60497 SRR957679.count
60497 SRR957680.count
241988 total
- 查看一下前四行
$ head -n 4 *.count
==> SRR957677.count <==
ENSG00000000003.14_2 804
ENSG00000000005.5_2 0
ENSG00000000419.12_2 379
ENSG00000000457.13_2 281
==> SRR957678.count <==
ENSG00000000003.14_2 350
ENSG00000000005.5_2 0
ENSG00000000419.12_2 173
ENSG00000000457.13_2 107
==> SRR957679.count <==
ENSG00000000003.14_2 786
ENSG00000000005.5_2 0
ENSG00000000419.12_2 397
ENSG00000000457.13_2 217
==> SRR957680.count <==
ENSG00000000003.14_2 949
ENSG00000000005.5_2 1
ENSG00000000419.12_2 506
ENSG00000000457.13_2 275
第一列ensembl_gene_id,第二列read_count计数
- 后4行
$ tail -n 4 *.count
==> SRR957677.count <==
__ambiguous 271271
__too_low_aQual 0
__not_aligned 1244591
__alignment_not_unique 7515355
==> SRR957678.count <==
__ambiguous 109197
__too_low_aQual 0
__not_aligned 560868
__alignment_not_unique 2996683
==> SRR957679.count <==
__ambiguous 289659
__too_low_aQual 0
__not_aligned 1303760
__alignment_not_unique 7943707
==> SRR957680.count <==
__ambiguous 327210
__too_low_aQual 0
__not_aligned 1407817
__alignment_not_unique 8772501
- 合并表达矩阵并进行注释(需要进入R环境)
查看原始数据,77和78是control,79和80是实验组
从上面看出需要至少做两步工作才能更好理解和往下进行分析
第一,需要把4个文件合并;
第二,需要把ensembl_gene_id转换为gene_symbol;(这一步不进行也行,后面还需要)
所以,上一步得到的4个单独的矩阵文件,现在要把这4个文件合并为行为基因名,列为样本名,中间为count的矩阵文件。
- 熟悉python的朋友可以参考这篇文章
- 我用R中的merge命令来处理,参考这里和这里
(1)载入数据,添加列名
> options(stringsAsFactors = FALSE)
> control1<-read.table("SRR957677.count",sep = "\t",col.names = c("gene_id","control1"))
> head(control1)
gene_id control1
1 ENSG00000000003.14_2 804
2 ENSG00000000005.5_2 0
3 ENSG00000000419.12_2 379
4 ENSG00000000457.13_2 281
5 ENSG00000000460.16_3 499
6 ENSG00000000938.12_2 0
> control2<-read.table("SRR957678.count",sep = "\t",col.names = c("gene_id","control2"))
> head(control2)
gene_id control2
1 ENSG00000000003.14_2 350
2 ENSG00000000005.5_2 0
3 ENSG00000000419.12_2 173
4 ENSG00000000457.13_2 107
5 ENSG00000000460.16_3 207
6 ENSG00000000938.12_2 0
> treat1<-read.table("SRR957679.count",sep = "\t",col.names = c("gene_id","treat1"))
> treat2<-read.table("SRR957680.count",sep = "\t",col.names = c("gene_id","treat1"))
(2)数据整合
- merge进行整合
- gencode的注释文件中的gene_id(如ENSMUSG00000105298.13_3)在EBI是不能搜索到的,所以用gsub功能只保留ENSMUSG00000105298这部分
处理签看一下最后五行
> tail(control1)
gene_id control1
60492 ENSG00000284600.1_1 0
60493 __no_feature 9022391
60494 __ambiguous 271271
60495 __too_low_aQual 0
60496 __not_aligned 1244591
60497 __alignment_not_unique 7515355
进行整合
> raw_count <- merge(merge(control1, control2, by="gene_id"), merge(treat1, treat2, by="gene_id"))
> head(raw_count)
gene_id control1 control2 treat1.x treat1.y
1 __alignment_not_unique 7515355 2996683 7943707 8772501
2 __ambiguous 271271 109197 289659 327210
3 __no_feature 9022391 3814709 8176659 10345267
4 __not_aligned 1244591 560868 1303760 1407817
5 __too_low_aQual 0 0 0 0
6 ENSG00000000003.14_2 804 350 786 949
> tail(raw_count)
gene_id control1 control2 treat1.x treat1.y
60492 ENSG00000284554.1_1 0 0 0 0
60493 ENSG00000284558.1_1 0 0 0 0
60494 ENSG00000284572.1_1 0 0 0 0
60495 ENSG00000284591.1_1 0 0 0 0
60496 ENSG00000284592.1_1 0 0 0 0
60497 ENSG00000284600.1_1 0 0 0 0
# 删除前五行,注意一下顺序。
> raw_count_filt <- raw_count[-1:-5,]
# 因为在EBI数据库中没办法找带小数点的基因,所以要替换成整数形式
# 第一步将匹配到的以及后面的数字连续匹配并替换为空,并赋值给ENSEMBL
> ENSEMBL <- gsub("\\.\\d*", "", raw_count_filt$gene_id)
> # 将ENSEMBL重新添加到raw_count_filt1矩阵
> row.names(raw_count_filt) <- ENSEMBL
> raw_count_filt <- cbind(ENSEMBL,raw_count_filt)
> colnames(raw_count_filt)[1] <- c("ensembl_gene_id")
> head(raw_count_filt)
(3)基因注释,获取gene_symbol
第一:去这里或这里的网页版,输入列表即可输出,不再赘述
第二:用bioMart对ensembl_id转换成gene_symbol
关于BiomaRt的安装,可以参考biomaRt包的安装
由于我输入install.packages("biomaRt")也同样出现错误,所以我按照上面的方法下载
# 先查看是否已安装openssl 若没安装则先进行安装
$ sudo apt-get install openssl
#安装这个的时候还蛮慢的
$ sudo apt-get install libssl-dev
# 安装这个的时候出现了问题(链接文章里,这个gnutls写错了!qaq)
$ sudo apt-get install libcurl4-gnutls-dev
然后我就在R里面,安装httr出现了问题

然后,关于解决这个问题的方法,单独发了一篇,R中安装httr出现的问题
感谢大佬带我,还有一个重新设置的问题,等我明天再发一篇叭。
$ R
>BiocManager::install("httr")
>BiocManager::install("XML")
>BiocManager::install("biomaRt")
#顺带一起把DESeq2安装了
>BiocManager::install('DESeq2')
十一点半了,我决定睡觉了。
明天继续
-------------------------------------分割线--------------------------------------------
下课回来了,我又来啦嘻嘻
#首先进入R环境
$ R
#载入BiomaRt和curl
> library('biomaRt')
> library(curl)
#因为还没学过R,所以只能照搬别人的代码了…(我学,我一定学!)
> mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
> my_ensembl_gene_id <- row.names(raw_count_filt)
> options(timeout = 4000000)
> hg_symbols<- getBM(attributes=c('ensembl_gene_id','hgnc_symbol',"chromosome_name", "start_position","end_position", "band"), filters= 'ensembl_gene_id', values = my_ensembl_gene_id, mart = mart)
加载完了之后,看一下
> head(hg_symbols)
ensembl_gene_id hgnc_symbol chromosome_name start_position end_position
1 ENSG00000003056 M6PR 12 8940361 8949761
2 ENSG00000081842 PCDHA6 5 140827958 141012347
3 ENSG00000105808 RASA4 7 102573807 102616757
4 ENSG00000116017 ARID3A 19 925781 975939
5 ENSG00000120235 IFNA6 9 21349835 21351378
6 ENSG00000123584 MAGEA9 X 149781930 149787737
band
1 p13.31
2 q31.3
3 q22.1
4 p13.3
5 p21.3
6 q28
将合并后的表达数据框raw_count_filt和注释得到的hg_symbols整合为一:
> readcount <- merge(raw_count_filt, hg_symbols, by="ensembl_gene_id")
> head(readcount)
ensembl_gene_id gene_id control1 control2 treat1.x treat1.y
1 ENSG00000003056 ENSG00000003056.3 662 280 591 732
2 ENSG00000081842 ENSG00000081842.13 0 0 0 0
3 ENSG00000105808 ENSG00000105808.13 0 0 4 2
4 ENSG00000116017 ENSG00000116017.6 276 108 270 336
5 ENSG00000120235 ENSG00000120235.3 0 0 0 0
6 ENSG00000123584 ENSG00000123584.7 0 0 0 0
hgnc_symbol chromosome_name start_position end_position band
1 M6PR 12 8940361 8949761 p13.31
2 PCDHA6 5 140827958 141012347 q31.3
3 RASA4 7 102573807 102616757 q22.1
4 ARID3A 19 925781 975939 p13.3
5 IFNA6 9 21349835 21351378 p21.3
6 MAGEA9 X 149781930 149787737 q28
输出count矩阵文件
> write.csv(readcount, file='readcount_all,csv')
> readcount<-raw_count_filt[ ,-1:-2]
> write.csv(readcount, file='readcount.csv')
> head(readcount)
control1 control2 treat1.x treat1.y
ENSG00000000003_2 804 350 786 949
ENSG00000000005_2 0 0 0 1
ENSG00000000419_2 379 173 397 506
ENSG00000000457_2 281 107 217 275
ENSG00000000460_3 499 207 440 538
ENSG00000000938_2 0 0 0 0
因为我主要参考的文章里有一处有点不一样,所以我主要参考的是
RNA-seq实战(中)_合并矩阵及DEseq2筛选差异并注释
备注:
因为我这里用到的是人样本测序数据,而教程里面都是小鼠,所以部分略有不同。 mart <- useDataset 用的是"hsapiens_gene_ensembl"
我这里的注释后,gene_id没有小数,所以ENSEMBL <- gsub("\.\d*", "", raw_count_filt$gene_id) 可操作可不操作,但是为了遵循流程,我还是按照教程一步步来。但是后面发现,合并时候出了很大问题,所以在前面操作中,我将第一列提取出来的ENSEMBL当做行名同时,还将其与数据合并cbind,并命名为enseble_gene_id,后面合并也是以这一列为准。否则,后面报错。
六、DESeq2筛选差异表达基因
1、载入数据(countData和colData)
> mycounts <- read.csv("readcount.csv")
> head(mycounts)
X control1 control2 treat1.x treat1.y
1 ENSG00000000003_2 804 350 786 949
2 ENSG00000000005_2 0 0 0 1
3 ENSG00000000419_2 379 173 397 506
4 ENSG00000000457_2 281 107 217 275
5 ENSG00000000460_3 499 207 440 538
6 ENSG00000000938_2 0 0 0 0
#这里有个x,需要去除,先把第一列当作行名来处理
> rownames(mycounts)<-mycounts[,1]
#把带X的列删除
> mycounts<-mycounts[,-1]
> head(mycounts)
control1 control2 treat1.x treat1.y
ENSG00000000003_2 804 350 786 949
ENSG00000000005_2 0 0 0 1
ENSG00000000419_2 379 173 397 506
ENSG00000000457_2 281 107 217 275
ENSG00000000460_3 499 207 440 538
ENSG00000000938_2 0 0 0 0
# 这一步很关键,要明白condition这里是因子,不是样本名称;小鼠数据有对照组和处理组,各两个重复
> condition <- factor(c(rep("control",2),rep("treat",2)), levels = c("control","treat"))
> condition
[1] control control treat treat
Levels: control treat
#colData也可以自己在excel做好另存为.csv格式,再导入即可
> colData <- data.frame(row.names=colnames(mycounts), condition)
> colData
condition
control1 control
control2 control
treat1.x treat
treat1.y treat
2、构建dds对象
> library("DESeq2")
> dds <- DESeqDataSetFromMatrix(mycounts, colData, design= ~ condition)
> dds <- DESeq(dds)
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
# 查看一下dds的内容
> dds
class: DESeqDataSet
dim: 60492 4
metadata(1): version
assays(4): counts mu H cooks
rownames(60492): ENSG00000000003_2 ENSG00000000005_2 ...
ENSG00000284592_1 ENSG00000284600_1
rowData names(22): baseMean baseVar ... deviance maxCooks
colnames(4): control1 control2 treat1.x treat1.y
colData names(2): condition sizeFactor
3、总体结果查看
> res = results(dds, contrast=c("condition", "control", "treat"))
> res = res[order(res$pvalue),]
> head(res)
log2 fold change (MLE): condition control vs treat
Wald test p-value: condition control vs treat
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE
ENSG00000178691_2 525.694712971668 2.89353494910036 0.236838344980599
ENSG00000135535_3 1179.93018699244 1.24009500894173 0.195787854442592
ENSG00000196504_3 1775.80082364858 1.12403000897508 0.201799837601116
ENSG00000141425_3 1300.90395042466 0.982783504147517 0.177217001121972
ENSG00000173905_2 545.305137710443 1.1832285473799 0.224038946461061
ENSG00000164172_3 254.782738750752 1.27726120796257 0.242907296366802
stat pvalue padj
ENSG00000178691_2 12.2173415345281 2.51164382359062e-34 2.16729745537635e-30
ENSG00000135535_3 6.33387097719766 2.39085467776778e-10 1.03153425072291e-06
ENSG00000196504_3 5.57002434856701 2.54703738492863e-08 6.31780309719697e-05
ENSG00000141425_3 5.5456502362948 2.92863743061628e-08 6.31780309719697e-05
ENSG00000173905_2 5.28135204200112 1.2823401858785e-07 0.000198215794614375
ENSG00000164172_3 5.25822495687344 1.45452524998957e-07 0.000198215794614375
> summary(res)
out of 30482 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 40, 0.13%
LFC < 0 (down) : 7, 0.023%
outliers [1] : 0, 0%
low counts [2] : 21853, 72%
(mean count < 124)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
#所有结果先进行输出
> write.csv(res,file="All_results.csv")
> table(res$padj<0.05)
FALSE TRUE
8597 32
注释:dds=DESeqDataSet Object
summary(res),一共40个genes上调,7个genes下调,没有离群值。padj小于0.05的共有32个。
4、提取差异表达genes(DEGs)
获取padj(p值经过多重校验校正后的值)小于0.05,表达倍数取以2为对数后大于1或者小于-1的差异表达基因。代码如下:
> diff_gene_deseq2 <-subset(res, padj < 0.05 & abs(log2FoldChange) > 1)
> dim(diff_gene_deseq2)
[1] 15 6
> head(diff_gene_deseq2)
log2 fold change (MLE): condition control vs treat
Wald test p-value: condition control vs treat
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE
ENSG00000178691_2 525.694712971668 2.89353494910036 0.236838344980599
ENSG00000135535_3 1179.93018699244 1.24009500894173 0.195787854442592
ENSG00000196504_3 1775.80082364858 1.12403000897508 0.201799837601116
ENSG00000173905_2 545.305137710443 1.1832285473799 0.224038946461061
ENSG00000164172_3 254.782738750752 1.27726120796257 0.242907296366802
ENSG00000172239_3 235.672693658546 1.33513402183508 0.254808834148659
stat pvalue padj
ENSG00000178691_2 12.2173415345281 2.51164382359062e-34 2.16729745537635e-30
ENSG00000135535_3 6.33387097719766 2.39085467776778e-10 1.03153425072291e-06
ENSG00000196504_3 5.57002434856701 2.54703738492863e-08 6.31780309719697e-05
ENSG00000173905_2 5.28135204200112 1.2823401858785e-07 0.000198215794614375
ENSG00000164172_3 5.25822495687344 1.45452524998957e-07 0.000198215794614375
ENSG00000172239_3 5.23974777521311 1.60796217673036e-07 0.000198215794614375
> write.csv(diff_gene_deseq2,file= "DEG_treat_vs_control.csv")
5、用BiomaRt对表达基因进行注释
> mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
> my_ensembl_gene_id<-row.names(diff_gene_deseq2)
> hg_symbols<- getBM(attributes=c('ensembl_gene_id','external_gene_name',"description"),
+ filters = 'ensembl_gene_id', values = my_ensembl_gene_id, mart = mart)
> head(hg_symbols)
ensembl_gene_id external_gene_name
1 ENSG00000128512 DOCK4
description
1 dedicator of cytokinesis 4 [Source:HGNC Symbol;Acc:HGNC:19192]
# 合并数据:res结果hg_symbols合并成一个文件
> head(diff_gene_deseq2)
log2 fold change (MLE): condition control vs treat
Wald test p-value: condition control vs treat
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE
ENSG00000178691_2 525.694712971668 2.89353494910036 0.236838344980599
ENSG00000135535_3 1179.93018699244 1.24009500894173 0.195787854442592
ENSG00000196504_3 1775.80082364858 1.12403000897508 0.201799837601116
ENSG00000173905_2 545.305137710443 1.1832285473799 0.224038946461061
ENSG00000164172_3 254.782738750752 1.27726120796257 0.242907296366802
ENSG00000172239_3 235.672693658546 1.33513402183508 0.254808834148659
stat pvalue padj
ENSG00000178691_2 12.2173415345281 2.51164382359062e-34 2.16729745537635e-30
ENSG00000135535_3 6.33387097719766 2.39085467776778e-10 1.03153425072291e-06
ENSG00000196504_3 5.57002434856701 2.54703738492863e-08 6.31780309719697e-05
ENSG00000173905_2 5.28135204200112 1.2823401858785e-07 0.000198215794614375
ENSG00000164172_3 5.25822495687344 1.45452524998957e-07 0.000198215794614375
ENSG00000172239_3 5.23974777521311 1.60796217673036e-07 0.000198215794614375
> ensembl_gene_id<-rownames(diff_gene_deseq2)
> diff_gene_deseq2<-cbind(ensembl_gene_id,diff_gene_deseq2)
> colnames(diff_gene_deseq2)[1]<-c("ensembl_gene_id")
> diff_name<-merge(diff_gene_deseq2,hg_symbols,by="ensembl_gene_id")
> head(diff_name)
DataFrame with 1 row and 9 columns
ensembl_gene_id baseMean log2FoldChange lfcSE
1 ENSG00000128512 179.89400102255 1.23959584130584 0.296973202603587
stat pvalue padj external_gene_name
1 4.17409998760226 2.99166354169603e-05 0.0135868761585763 DOCK4
description
1 dedicator of cytokinesis 4 [Source:HGNC Symbol;Acc:HGNC:19192]
> CD164 <- diff_name[diff_name$external_gene_name=="CD164",]
> CD164
DataFrame with 0 rows and 9 columns
至此,差异表达基因提取并注释完毕,下一步
- 先进行数据可视化(Data visulization)
- 然后进行富集分分析及可视化
七、数据可视化
1、MA plot
> plotMA(res,ylim=c(-2,2))
> topGene <- rownames(res)[which.min(res$padj)]
> with(res[topGene, ], {
+ points(baseMean, log2FoldChange, col="dodgerblue", cex=6, lwd=2)
+ text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
+ })
结果如下:
经过lfcShrink 收缩log2 fold change
> res_order<-res[order(row.names(res)),]
> res = res_order
> res.shrink <- lfcShrink(dds, contrast = c("condition","treat","control"), res=res)
using 'normal' for LFC shrinkage, the Normal prior from Love et al (2014).
Note that type='apeglm' and type='ashr' have shown to have less bias than type='normal'.
See ?lfcShrink for more details on shrinkage type, and the DESeq2 vignette.
Reference: https://doi.org/10.1093/bioinformatics/bty895
> plotMA(res.shrink, ylim = c(-5,5))
> topGene <- rownames(res)[which.min(res$padj)]
> with(res[topGene, ], {
+ points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
+
+ text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
+ })
> plotMA(res.shrink, ylim = c(-5,5))
> topGene <- rownames(res)[which.min(res$padj)]
> with(res[topGene, ], {
+ points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
+ text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
+ })
结果如下:
2、Plot counts
DESeq2提供了一个plotCounts()函数来查看某一个感兴趣的gene在组间的差别。counts会根据groups分组。更多的参数请输入命令?plotCounts
后面的暂时不写了qaq感兴趣可以看
探索分析结果:Data visulization