进阶版Venn plot:Upset plot入门实战代码详解——UpSetR包介绍
网上已有一些帖子讲解了Upset plot的背景知识和实现方法,本文旨在从实战角度出发,解读Upset plot实现的一些途径及其优劣势以及如何通过upsetR包实现Upset plot的绘制,并提供代码和input格式示例。另博主在使用中发现一处报错,尚不清楚原因,一并提出,以抛砖引玉。
Q1:Upset plot和Venn plot的关系?
提到集合的可视化,传统的方法以Venn plot为主。然而当集合的数量过多(超过五个),Venn plot会变得非常杂乱(Fig.1)。Upset plot可以很好地展示多个集合之间的关系(Fig.2)。两种图的样子虽然差别较大,但是其本质都是反映了集合的关系。简而言之有三点:
- 集合数<5使用Venn plot更加清晰。
- 集合数>5使用Upset plot更加清晰。
- Upset plot具有更多的展示方式。
 (Fig.1)
(Fig.1)
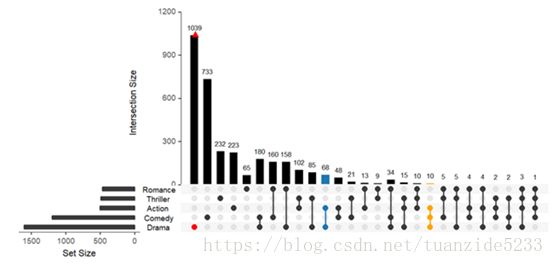 (Fig.2)
(Fig.2)
Q2:如何解读Upset plot?
熟悉Venn plot的朋友只需稍作变换即可很好地理解Upset plot。Fig.2是一个简单的Upset plot,图像可以分为上部分(条形图)和下部分(左侧的条形图,中间的集合名称和右侧的点阵)。左下方的横行条形图展示了每个集合的元素数(此处元素数由input文件直接得到),左中方的集合名称由input文件中命名的集合名得到,左下方的点阵应与上方的条形图一起解读。有颜色的点代表涉及到相关集合,即有颜色的点对应的左中的集合一起取交集。以红点为例,代表Drama与Drama集合取交集,即自身取交集。蓝点代表Action集合与Drama取交集,对应的条形图数值为68,即有68个元素既在Action中又在Drama中。以此类推便可理解黄点。上方的条形图代表着每个交集对应的元素数。
Q3:如何绘制Upset plot?
Upset plot的绘制方法大致有三种:
1.通过网站绘制
有一些网站支持绘制各种统计图,此处推荐一个:易汉博生物信息在线作图(http://www.ehbio.com/ImageGP/index.php/Home/Index/UpsetView.html)。该网站囊括了生物信息学领域所需要的多种基础图像。
优点:简单易用;缺点:可编辑性低,不能满足复杂的显示要求,大样本量时不稳定
2. 通过软件实现
TBtools是一款旨在帮助生命科学领域科研人员实现无代码数据分析及绘图的软件。关于这个软件的使用详情可见:https://mp.weixin.qq.com/s/MPlN2LtsCn3FqEM2ZkLa5A
优缺点同上。
3.使用UpSetR包绘图
UpSetR包是基于R语言开发的一款程序包,可以用于多领域的集合可视化。
优点:可编辑性好,可以满足大样本分析需求;缺点:R语言使用门槛高于前两种方式
网友(ID:严涛)和网友(ID:hoptop)分别在51CTO平台(http://bigdata.51cto.com/art/201710/554293.htm)和简书(https://www.jianshu.com/p/324aae3d5ea4)发表了以UpSetR包提供的样本为基础的程序讲解,文章详细阐述了各种参数的意义。然而对于初学者来说,其中涉及到的一些操作属于锦上添花,增加了文章的理解难度。本文将尽可能的减少代码量,使用通俗易懂的语言,以便于理解和重复。
一、数据准备
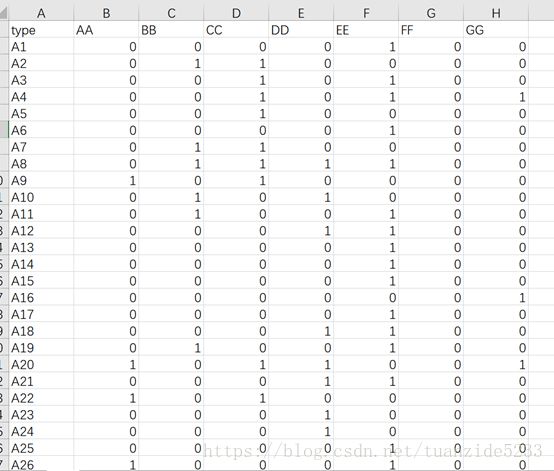 (Fig.3)
(Fig.3)
制作如Fig.3所示的矩阵,列名称为集合名称,行名称为元素名称,0表示不在该集合,1表示在该集合,为了便于后续操作,将文件保存为csv格式。
二、安装UpSetR包
source("http://bioconductor.org/biocLite.R")
biocLite("UpSetR")三、导入矩阵
library(UpSetR)
setwd("E:/example")
example = read.csv("example.csv",header=TRUE,row.names=1,check.names = FALSE)
knitr::kable(head(example[,1:7]))第一行:载入程序包
第二行:定义矩阵所在路径
第三行:将example.csv文件中的矩阵读取并存储到example变量中
第四行:预览矩阵的前7列
四、绘制Upset plot
upset(example, sets = c("AA", "BB", "CC", "DD", "EE",
"FF", "GG"), mb.ratio = c(0.55, 0.45), order.by = "freq",
queries = list(list(query=intersects, params=list("CC", "EE"), color="red", active=T),
list(query=intersects, params=list("CC", "DD", "EE"), color="green", active=T),
list(query=intersects, params=list("CC", "DD", "EE", "AA"), color="blue", active=T)),
nsets = 7, number.angles = 0, point.size = 6, line.size = 1, mainbar.y.label = "title",
sets.x.label = "title", text.scale = c(2, 2, 2, 2, 2, 2))
 (Fig.4)
(Fig.4)
example:存放矩阵的变量名称
set:所需要的集合名称
mb.ratio:调整上下两部分的比例
order.by:排序方式,freq为按频率排序
queries:查询函数,用于对指定列添加颜色,结合代码可知分别为CC&EE/CC&DD&EE/AA&CC&DD&EE列添加了红色、绿色和蓝色
active:如果为F,则在每一列上方显示一个三角(Fig.5)
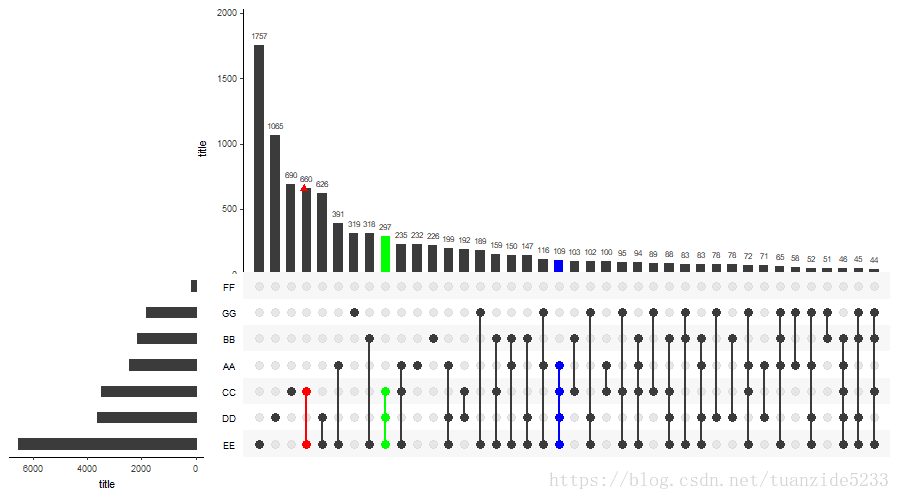
(Fig.5)
nset:集合数量,也可用set参数指定具体集合
number.angles:上方条形图数字角度,0为横向,90为竖向,但90时不在正上方
point.size:下方点阵中点的大小
line.size:下方点阵中每个线的粗细
mainbar.y.label:上方条形图Y轴名称
sets.x.label:左下方条形图X轴名称
text.scale:六个数字控制关系见Fig.6

UpSetR包的介绍就到此为止了,还有更加丰富的功能可参考网友(ID:严涛)和网友(ID:hoptop)的文章。
在实际使用中,博主发现:

(Fig.7)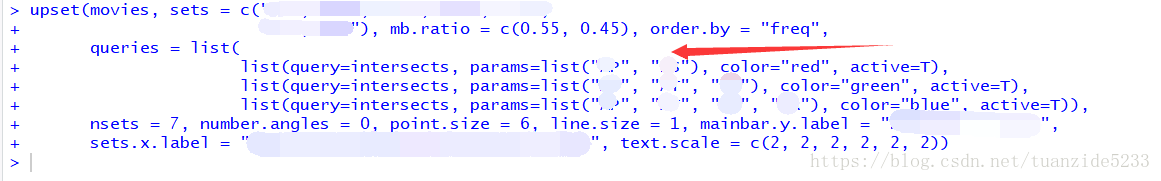
(Fig.8)
Fig.7是报错的,Fig.8代码中删掉了Fig.7中的只有单个集合的那一行,就不会提醒列不匹配了。换句话说只要是params的list包括两个或两个以上就不报错。换了一个版本的R仍然存在这个问题。尚不清楚原因,欢迎留言讨论。
GEO芯片数据差异表达分析时需要log2处理的原因
https://blog.csdn.net/tuanzide5233/article/details/88542805
GEO芯片数据差异表达分析时是否需要log2以及标准化的问题
https://blog.csdn.net/tuanzide5233/article/details/88542558
差异表达矩阵制作教程
https://blog.csdn.net/tuanzide5233/article/details/83659768
差异表达的热图绘制详见
https://blog.csdn.net/tuanzide5233/article/details/83659501
使用edgeR对RNAseq数据进行差异表达分析教程
https://blog.csdn.net/tuanzide5233/article/details/88785486
差异表达分析(DEG)时 row.names'里不能有重复的名字 的解决方案
https://blog.csdn.net/tuanzide5233/article/details/86568155
生存分析系列教程(一)使用生信人工具盒进行生存分析
https://blog.csdn.net/tuanzide5233/article/details/83685403
富集分析与蛋白质互作用网络(PPI)的可视化 Cystocape入门指南
https://blog.csdn.net/tuanzide5233/article/details/88048439
进阶版Venn plot:Upset plot入门实战代码详解——UpSetR包介绍
https://blog.csdn.net/tuanzide5233/article/details/83109527
使用R语言ggplot2包绘制pathway富集分析气泡图(Bubble图):数据结构及代码
https://blog.csdn.net/tuanzide5233/article/details/82141817