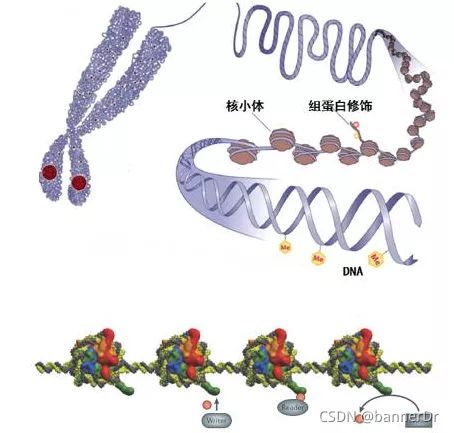
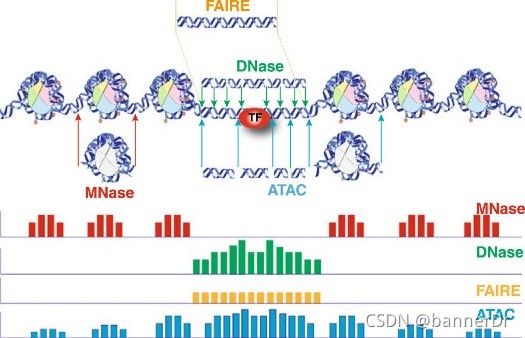
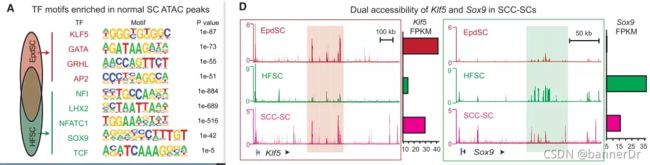
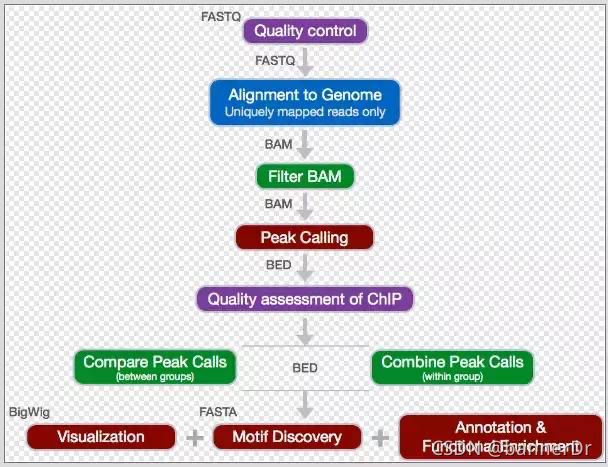
- Signac::EnhanceCoveragePlot 参考实现流程
倪桦
r语言Signaccoverageplot
Signac中的CoveragePlot是一种用于展示基因组覆盖度的图形工具,常用于ATAC-seq(AssayforTransposase-AccessibleChromatinusingsequencing)数据分析。它显示了特定基因组区域内测序读取的覆盖度,即每个位置上读取的频率。覆盖度图形对于理解基因组的开放区域、调控元件活性以及染色质状态等方面具有重要作用。通过将多个样本的Coverag
- Week26 — 人类原发性肿瘤的染色质可及性图谱-03
六六_ryx
Week24—人类原发性肿瘤的染色质可及性图谱-01:主要回顾了ATAC-seq方法的原理和优点,并与其他研究染色质可及性方法的比较,然后介绍了这篇文章的主要结果和亮点以及提供的数据资源。Week25—人类原发性肿瘤的染色质可及性图谱-02:介绍了文章思路和主要结果。这篇文章主要了解下补充材料的分析方法。1.ATAC-seq数据预处理和比对ATAC-seq预处理和比对使用的是PEPATACpipe
- deeptools工具系列——比较不同bw_bam的信号差异
Z_bioinfo
目录前言bamCompare工具1.1工具原理1.2使用说明1.3使用实例bigwigCompare工具2.1使用说明2.2使用实例前言今天主要介绍下,如果我有2个bw/bam文件,我想要比较这2个文件信号之间的差异时,要如何进行分析。这个听上去是不是很像差异分析?对的,其实就是差异分析的简化版。而这个的实际用途,可以是:分别有实验组和对照组ATAC-seq的bam/bw文件,此时我们可以直接通过
- ATAC-seq发篇测序文章就结束了吗?看如何利用ATAC-seq数据为后续关键基因的转录调控研究提供重要依据
爱基百客
学习ATAC转录调控
染色质可及性(ChromatinAccessibility)是染色质的一种特性,为转录因子结合靶基因提供了空间。转座酶可及染色质测序分析(ATAC-seq)是常见的研究染色质可及性的方法,ATAC-seq联合RNA-seq是一种新的研究思路,为阐述基因组和特定生物过程中的基因差异表达提供见解。然而,做完ATAC-seq仅仅是发一篇测序类的文章吗?ATAC-seq能为后续的研究提供些什么?2024年
- 全反式维甲酸(ATAR)诱导白血细胞的染色质构象改变而影响分化进程
d5c9c866171f
AlterationsofspecificchromatinconformationaffectATRA-inducedleukemiacelldifferentiation期刊:CellDeath&Disease影响因子:6.044主要技术:Hi-C、ATAC-seq、ChIP-seq、RNA-seq发表时间:2018年研究背景染色体形成多层次构象在基因表达调控中起着至关重要的作用。高通量染色体
- 使用ArchR分析单细胞ATAC-seq数据(第十三章)
xuzhougeng
本文首发于我的个人博客,http://xuzhougeng.top/往期回顾:使用ArchR分析单细胞ATAC-seq数据(第一章)使用ArchR分析单细胞ATAC-seq数据(第二章)使用ArchR分析单细胞ATAC-seq数据(第三章)使用ArchR分析单细胞ATAC-seq数据(第四章)使用ArchR分析单细胞ATAC-seq数据(第五章)使用ArchR分析单细胞ATAC-seq数据(第六章
- ATAC-seq分析:Peak Calling(8)
冷冻工厂
1.寻找开发区域ATACseq的一个共同目标是识别转录因子结合和/或转录机制活跃的无核小体区域。该核小体游离信号对应于小于一个核小体的片段(如Greenleaf论文中定义<100bp)。然而,为了识别开放的染色质,我们可以简单地使用在测序中正确配对的所有读数(<2000bp)。2.无核小体区域有许多方法可用于从ATACseq数据中调用无核小体区域,其中许多方法借鉴自ChIPseq分析。一种非常流行
- Nature Commun|ATAC-seq探究复发性小儿B系急性淋巴细胞白血病的染色质可及性图谱
爱基百客
ATAC其他
表观基因组学分析是解释非编码基因组功能的重要方法之一。表观基因组学特征作为肿瘤细胞的基本特征,对发病机制、临床行为和治疗具有影响。在所有表观基因组标记中,组蛋白修饰和DNA甲基化已得到最广泛的研究,以深入了解表观基因组失调。染色质可及性是DNA调控元件的一个标志,新出现的证据表明它在癌症中发挥着重要作用。使用测序(ATAC-seq)检测转座酶可及染色质的方法的出现和优化使得在原发性癌症的全基因组范
- 从CNS封面文章中找数据 | Science:人类原发性肿瘤染色质可及性图谱
尐尐呅
今天给大家分享的ATAC-seq数据资源来自2018年的Science封面文章,美国斯坦福大学研究人员绘制了23种癌症全基因组染色质可及性图谱,为癌症研究提供基础数据资源。利用ATAC-seq对来自TCGA的涉及23种人类癌症类型的410个肿瘤样本进行染色质可及性分析,鉴定了近56.3万个DNA调节元件,绘制了796个全基因组染色质可及性图谱。为什么要绘制癌症染色质可及性图谱?癌症是全球死亡的主要
- 单细胞从理论到实践-这一本书就够了
生信交流平台
前面给大家介绍过一些单细胞相关的理论和实践知识BD单细胞测序平台Rhapsody(视频)10X单细胞RNA-seq技术(视频)10X单细胞混样技术(视频)单细胞ATAC-seq技术介绍免疫组库10XGenomicsVDJ测序10XGenomics的Visium空间基因表达解决方案空间转录组学(SpatialTranscriptomics)单细胞数据分析相关R包安装(视频)单细胞小提琴图+箱型图Se
- 软件9 —— MACS2
果蝇饲养员的生信笔记
一、基本介绍※峰识别(peakcalling)工具包括:MACS2和HOMER(适用于ChIP-seq和ATAC-seq)、HMMRATAC(针对于ATAC-seq)、exomePeak(针对m6A-seq)。※差异峰分析(peakdifferentialanalysis)工具包括:HOMER、DiffBind,它们是consensuspeak-based工具,假设数据分布是负二项分布。conse
- BED文件与bedtools简介
筱贺学生信
生信python开发语言
1、什么是bed格式1、文本文件2、表明基因组的一段区域3、标准的bed文件最少三列,最多十二列eg:1、chrom孔2、start开始3、end结束4、name名称5、score存一个数6、strand+or-2、bed格式的使用1、储存基因区2、储存基因组的某些位点信息3、储存CHIP-seq、ATAC-seq等的富集的peak信息3、bedtools是一种常用的bed操作工具,可以实现非常多
- 使用ArchR分析单细胞ATAC-seq数据(第五章)
xuzhougeng
本文首发于我的个人博客,http://xuzhougeng.top/往期回顾:使用ArchR分析单细胞ATAC-seq数据(第一章)使用ArchR分析单细胞ATAC-seq数据(第二章)使用ArchR分析单细胞ATAC-seq数据(第三章)使用ArchR分析单细胞ATAC-seq数据(第四章)第5章:使用ArchR聚类大部分单细胞聚类算法都在降维后空间中计算最近邻图,然后鉴定"社区"或者细胞聚类。
- ATAC-seq经典之作
Shaoqian_Ma
论文标题:Transpositionofnativechromatinforfastandsensitiveepigenomicprofilingofopenchromatin,DNA-bindingproteinsandnucleosomeposition表观测序领域大牛WilliamJGreenleaf和HowardYChang的经典之作,引用上千次的ATAC经典原文概述通过加上adaptor
- Cell Reports | 表观组学和单细胞测序揭示在急性应激条件下FoxM1协调β细胞亚群的细胞分裂、蛋白质合成和线粒体活性
爱基百客
单细胞测序ATAC其他
发表单位:瑞士分子健康科学研究所期刊:CellReports(IF:8.8)发表日期:2023年8月29日研究技术:ATAC-seq、ChIP-seq、RNA-seq、scRNA-seq(爱基百客均可以提供)2023年8月,瑞士分子健康科学研究所的研究团队在CellReports期刊(IF=8.8)上发表了题为“FoxM1coordinatescelldivision,proteinsynthes
- 使用ArchR分析单细胞ATAC-seq数据(第十四章)
xuzhougeng
本文首发于我的个人博客,http://xuzhougeng.top/往期回顾:使用ArchR分析单细胞ATAC-seq数据(第一章)使用ArchR分析单细胞ATAC-seq数据(第二章)使用ArchR分析单细胞ATAC-seq数据(第三章)使用ArchR分析单细胞ATAC-seq数据(第四章)使用ArchR分析单细胞ATAC-seq数据(第五章)使用ArchR分析单细胞ATAC-seq数据(第六章
- Plant Cell | DAP-seq和ATAC-seq助力构建玉米胚乳发育调控网络
爱基百客
DAP-seq其他
禾本科植物,如水稻(Oryzasativa)和玉米(Zeamays)的胚乳,不仅是种子发芽和幼苗早期生长的能量来源,也是人类食物、畜禽饲料及工业用途的重要原料。胚乳的发展经历两个主要阶段:一是细胞发展阶段,此时细胞会经历大量的分裂与分化;二是籽粒灌浆阶段,细胞在此期间积累淀粉和蛋白质等储存物质。三个关键转录因子,即NAKEDENDOSPERM1(NKD1)、NKD2和OPAQUE2(O2),在玉米
- ATAC-seq分析干货-2
生信阿拉丁
作者|Arno审稿|童蒙编辑|amethyst上一期我们介绍了ATAC-seq相关的背景知识。ATAC-Seq能帮助我们从全基因组范围内推测可能的开放染色质位点,其分析从本质上来说和ChIP-seq区别不大,核心都是peak-calling。如果现在你还不太了解基本分析流程,那快快跟随小编继续学起来吧。分析步骤前期我们通过Trimmomatic软件对原始下机数据进行了数据清洗,主要包括:去除下机数
- 使用ArchR分析单细胞ATAC-seq数据(第六章)
xuzhougeng
本文首发于我的个人博客,http://xuzhougeng.top/往期回顾:使用ArchR分析单细胞ATAC-seq数据(第一章)使用ArchR分析单细胞ATAC-seq数据(第二章)使用ArchR分析单细胞ATAC-seq数据(第三章)使用ArchR分析单细胞ATAC-seq数据(第四章)使用ArchR分析单细胞ATAC-seq数据(第五章)第6章:单细胞嵌入在ArchR中,类似于UMAP和t
- ATAC-seq专题---生信分析流程
诺禾致源
ATAC-seq信息分析流程主要分为以下几个部分:数据质控、序列比对、峰检测、motif分析、峰注释、富集分析,下面将对各部分内容进行展开讲解。一、测序数据过滤与质量评估下机数据经过过滤去除接头含量过高或低质量的reads,得到cleanreads用于后续分析。常见的trim软件有Trimmomatic、Skewer、fastp等。fastp是一款比较新的软件,使用时可以用--adapter_se
- ATACseq--染色质开放性测序
HOLLYxiao
ATAC-seq:利用转座酶研究染色质可进入性的高通量测序技术染色体/质结构:细胞核DNA与组蛋白相结合,DNA缠绕在组蛋白上形成串珠式结构,因此染色质结构高度折叠(压缩)。Tn5转座酶预先载入测序接头,在切除DNA的同时对DNA片段进行标记,ATAC-seq就是通过用Tn5转座酶探测开放的染色质来鉴定可接近的DNA区域。纯化标记的DNA片段,通过PCR扩增并测序。然后可以使用测序reads来推断
- ATAC-Seq与ChIP-Seq的异同
RachaelRiggs
ATAC-Seq与ChIP-Seq的异同ATAC-Seq与ChIP-Seq的不同的是ATAC-Seq是全基因组范围内检测染色质的开放程度,可以得到全基因组范围内的蛋白质可能结合的位点信息,一般用于不知道特定的转录因子,用此方法与其他方法结合筛查感兴趣的特定调控因子;但是ChIP-Seq是明确知道感兴趣的转录因子是什么,根据感兴趣的转录因子设计抗体去做ChIP实验拉DNA,验证感兴趣的转录因子是否与
- 单细胞ATAC和RNA测序的结合分析
概普生信
大家好,今天给大家分享的文章是今年9月发表在NatureCommunications(IF:14.919)上面的一篇文章。前列腺癌具有异质性,那些对系统治疗有反应的患者,如果存在某些方法对这些患者进行分层,那将使患者受益。这篇文章就是采用ATAC-seq和RNA-seq的单细胞检测方法,对恩杂鲁胺(一种抗癌药物)的早期治疗反应和耐药性模型进行研究。方法RNA测序及预处理LNCaP和VCaP细胞系是
- Molecular Cancer|CDK9抑制诱导表观遗传重编程,揭示了规避淋巴瘤耐药性的策略
爱基百客
ATAC单细胞ChIP其他
细胞周期蛋白依赖性激酶(CDK)蛋白家族在细胞周期进程(如CDK1/2/4/6)和RNA转录(如CDK7/8/9/11)的调控中起着不可或缺的作用。由于染色体区域易位或基因扩增导致的CDKs表达失调与肿瘤发生有关。在淋巴瘤细胞中,如何通过ATAC-seq和ChIP-seq揭示CDK9抑制诱导表观遗传重编程?看看今天带来的这个文章。发表单位:美国希望之城国家医疗中心期刊:MolecularCance
- Week7—TFmapper: 用ChIP-seq/ATAC-seq/DNase-seq数据预测感兴趣基因的调控因子
六六_ryx
原文:TFmapper:AToolforSearchingPutativeFactorsRegulatingGeneExpressionUsingChIP-seqDataDOI:10.7150/ijbs.28850发表日期:2018.09.07作者:JianmingZeng(大神哦)TFmapper:http://www.tfmapper.org/功能:用于搜索感兴趣的基因的调控因子感兴趣基因组区
- ATAC-seq数据可视化——超简单教程
基因组学研究生
大家好这里是有时候季更,有时候月更的基因组学研究生。上一期记录了ATAC数据从Fastq数据到bam文件的处理脚本。ATAC数据一键处理基因组学研究生,公众号:基因组学研究生一个不成熟的小脚本,ATAC数据一键预处理从fastq到treatedbam后来看到有同学后台问我有没有ATAC-seq可视化教程,所以简单写了写,希望能帮到部分人~Ps:那位问我的同学,由于我消息晚了几天看到,所以回复不了了
- 秘籍 | 计算基因组的有效大小(effective genome size)
生信卷王
写在前面最近在ATAC-Seq中callpeaks的时候发现需要用到基因组的有效大小(effectivegenomesize)方法一自己编写脚本#coding=utf-8importsysaList=[]fa_file=sys.argv[1]withopen(fa_file,'r')asf:forlineinf:line=line.strip()line=line.upper()ifnotline
- GenomicRanges 根据重合比例合并基因区间数据
BeeBee生信
最近在做ATAC-Seq分析,得到实验组和对照组peaksets(本文称为peaks1和peaks2)后想合并得到consensuspeaksets.用bedtoolsintersect做可以设置重合比例参数,但它无法从peaks1和peaks2计算并集为新peaksets然后输出。方法二是自己写python代码,但我觉得GenomicRanges可以实现,没必要重复造轮子。实现过程大致如下:先f
- [生信]单细胞测序数据分析的各类软件汇总
MC学公卫
Github上有人收录了各类单细胞RNA测序的数据分析软件,包括RNA-seq和ATAC-seq等。https://github.com/seandavi/awesome-single-cell
- ATAC-seq总结
otw_5f1c
背景知识什么是ATAC-Seq?Assayfortransposase-accessiblechromatinusingsequencing(利用转座酶研究染色质可接近性的高通量测序技术),可以在全基因上捕获裸露DAN与核小体的DNA。细胞的不同时期染色质松紧程度不同,其中结构疏松的染色质被称为开放染色质,它有足够的区域允许一些调控蛋白过来与其结合。染色质的这种特性,叫做染色质的可接近性。ATAC
- java封装继承多态等
麦田的设计者
javaeclipsejvmcencapsulatopn
最近一段时间看了很多的视频却忘记总结了,现在只能想到什么写什么了,希望能起到一个回忆巩固的作用。
1、final关键字
译为:最终的
&
- F5与集群的区别
bijian1013
weblogic集群F5
http请求配置不是通过集群,而是F5;集群是weblogic容器的,如果是ejb接口是通过集群。
F5同集群的差别,主要还是会话复制的问题,F5一把是分发http请求用的,因为http都是无状态的服务,无需关注会话问题,类似
- LeetCode[Math] - #7 Reverse Integer
Cwind
java题解MathLeetCodeAlgorithm
原题链接:#7 Reverse Integer
要求:
按位反转输入的数字
例1: 输入 x = 123, 返回 321
例2: 输入 x = -123, 返回 -321
难度:简单
分析:
对于一般情况,首先保存输入数字的符号,然后每次取输入的末位(x%10)作为输出的高位(result = result*10 + x%10)即可。但
- BufferedOutputStream
周凡杨
首先说一下这个大批量,是指有上千万的数据量。
例子:
有一张短信历史表,其数据有上千万条数据,要进行数据备份到文本文件,就是执行如下SQL然后将结果集写入到文件中!
select t.msisd
- linux下模拟按键输入和鼠标
被触发
linux
查看/dev/input/eventX是什么类型的事件, cat /proc/bus/input/devices
设备有着自己特殊的按键键码,我需要将一些标准的按键,比如0-9,X-Z等模拟成标准按键,比如KEY_0,KEY-Z等,所以需要用到按键 模拟,具体方法就是操作/dev/input/event1文件,向它写入个input_event结构体就可以模拟按键的输入了。
linux/in
- ContentProvider初体验
肆无忌惮_
ContentProvider
ContentProvider在安卓开发中非常重要。与Activity,Service,BroadcastReceiver并称安卓组件四大天王。
在android中的作用是用来对外共享数据。因为安卓程序的数据库文件存放在data/data/packagename里面,这里面的文件默认都是私有的,别的程序无法访问。
如果QQ游戏想访问手机QQ的帐号信息一键登录,那么就需要使用内容提供者COnte
- 关于Spring MVC项目(maven)中通过fileupload上传文件
843977358
mybatisspring mvc修改头像上传文件upload
Spring MVC 中通过fileupload上传文件,其中项目使用maven管理。
1.上传文件首先需要的是导入相关支持jar包:commons-fileupload.jar,commons-io.jar
因为我是用的maven管理项目,所以要在pom文件中配置(每个人的jar包位置根据实际情况定)
<!-- 文件上传 start by zhangyd-c --&g
- 使用svnkit api,纯java操作svn,实现svn提交,更新等操作
aigo
svnkit
原文:http://blog.csdn.net/hardwin/article/details/7963318
import java.io.File;
import org.apache.log4j.Logger;
import org.tmatesoft.svn.core.SVNCommitInfo;
import org.tmateso
- 对比浏览器,casperjs,httpclient的Header信息
alleni123
爬虫crawlerheader
@Override
protected void doGet(HttpServletRequest req, HttpServletResponse res) throws ServletException, IOException
{
String type=req.getParameter("type");
Enumeration es=re
- java.io操作 DataInputStream和DataOutputStream基本数据流
百合不是茶
java流
1,java中如果不保存整个对象,只保存类中的属性,那么我们可以使用本篇文章中的方法,如果要保存整个对象 先将类实例化 后面的文章将详细写到
2,DataInputStream 是java.io包中一个数据输入流允许应用程序以与机器无关方式从底层输入流中读取基本 Java 数据类型。应用程序可以使用数据输出流写入稍后由数据输入流读取的数据。
- 车辆保险理赔案例
bijian1013
车险
理赔案例:
一货运车,运输公司为车辆购买了机动车商业险和交强险,也买了安全生产责任险,运输一车烟花爆竹,在行驶途中发生爆炸,出现车毁、货损、司机亡、炸死一路人、炸毁一间民宅等惨剧,针对这几种情况,该如何赔付。
赔付建议和方案:
客户所买交强险在这里不起作用,因为交强险的赔付前提是:“机动车发生道路交通意外事故”;
如果是交通意外事故引发的爆炸,则优先适用交强险条款进行赔付,不足的部分由商业
- 学习Spring必学的Java基础知识(5)—注解
bijian1013
javaspring
文章来源:http://www.iteye.com/topic/1123823,整理在我的博客有两个目的:一个是原文确实很不错,通俗易懂,督促自已将博主的这一系列关于Spring文章都学完;另一个原因是为免原文被博主删除,在此记录,方便以后查找阅读。
有必要对
- 【Struts2一】Struts2 Hello World
bit1129
Hello world
Struts2 Hello World应用的基本步骤
创建Struts2的Hello World应用,包括如下几步:
1.配置web.xml
2.创建Action
3.创建struts.xml,配置Action
4.启动web server,通过浏览器访问
配置web.xml
<?xml version="1.0" encoding="
- 【Avro二】Avro RPC框架
bit1129
rpc
1. Avro RPC简介 1.1. RPC
RPC逻辑上分为二层,一是传输层,负责网络通信;二是协议层,将数据按照一定协议格式打包和解包
从序列化方式来看,Apache Thrift 和Google的Protocol Buffers和Avro应该是属于同一个级别的框架,都能跨语言,性能优秀,数据精简,但是Avro的动态模式(不用生成代码,而且性能很好)这个特点让人非常喜欢,比较适合R
- lua set get cookie
ronin47
lua cookie
lua:
local access_token = ngx.var.cookie_SGAccessToken
if access_token then
ngx.header["Set-Cookie"] = "SGAccessToken="..access_token.."; path=/;Max-Age=3000"
end
- java-打印不大于N的质数
bylijinnan
java
public class PrimeNumber {
/**
* 寻找不大于N的质数
*/
public static void main(String[] args) {
int n=100;
PrimeNumber pn=new PrimeNumber();
pn.printPrimeNumber(n);
System.out.print
- Spring源码学习-PropertyPlaceholderHelper
bylijinnan
javaspring
今天在看Spring 3.0.0.RELEASE的源码,发现PropertyPlaceholderHelper的一个bug
当时觉得奇怪,上网一搜,果然是个bug,不过早就有人发现了,且已经修复:
详见:
http://forum.spring.io/forum/spring-projects/container/88107-propertyplaceholderhelper-bug
- [逻辑与拓扑]布尔逻辑与拓扑结构的结合会产生什么?
comsci
拓扑
如果我们已经在一个工作流的节点中嵌入了可以进行逻辑推理的代码,那么成百上千个这样的节点如果组成一个拓扑网络,而这个网络是可以自动遍历的,非线性的拓扑计算模型和节点内部的布尔逻辑处理的结合,会产生什么样的结果呢?
是否可以形成一种新的模糊语言识别和处理模型呢? 大家有兴趣可以试试,用软件搞这些有个好处,就是花钱比较少,就算不成
- ITEYE 都换百度推广了
cuisuqiang
GoogleAdSense百度推广广告外快
以前ITEYE的广告都是谷歌的Google AdSense,现在都换成百度推广了。
为什么个人博客设置里面还是Google AdSense呢?
都知道Google AdSense不好申请,这在ITEYE上也不是讨论了一两天了,强烈建议ITEYE换掉Google AdSense。至少,用一个好申请的吧。
什么时候能从ITEYE上来点外快,哪怕少点
- 新浪微博技术架构分析
dalan_123
新浪微博架构
新浪微博在短短一年时间内从零发展到五千万用户,我们的基层架构也发展了几个版本。第一版就是是非常快的,我们可以非常快的实现我们的模块。我们看一下技术特点,微博这个产品从架构上来分析,它需要解决的是发表和订阅的问题。我们第一版采用的是推的消息模式,假如说我们一个明星用户他有10万个粉丝,那就是说用户发表一条微博的时候,我们把这个微博消息攒成10万份,这样就是很简单了,第一版的架构实际上就是这两行字。第
- 玩转ARP攻击
dcj3sjt126com
r
我写这片文章只是想让你明白深刻理解某一协议的好处。高手免看。如果有人利用这片文章所做的一切事情,盖不负责。 网上关于ARP的资料已经很多了,就不用我都说了。 用某一位高手的话来说,“我们能做的事情很多,唯一受限制的是我们的创造力和想象力”。 ARP也是如此。 以下讨论的机子有 一个要攻击的机子:10.5.4.178 硬件地址:52:54:4C:98
- PHP编码规范
dcj3sjt126com
编码规范
一、文件格式
1. 对于只含有 php 代码的文件,我们将在文件结尾处忽略掉 "?>" 。这是为了防止多余的空格或者其它字符影响到代码。例如:<?php$foo = 'foo';2. 缩进应该能够反映出代码的逻辑结果,尽量使用四个空格,禁止使用制表符TAB,因为这样能够保证有跨客户端编程器软件的灵活性。例
- linux 脱机管理(nohup)
eksliang
linux nohupnohup
脱机管理 nohup
转载请出自出处:http://eksliang.iteye.com/blog/2166699
nohup可以让你在脱机或者注销系统后,还能够让工作继续进行。他的语法如下
nohup [命令与参数] --在终端机前台工作
nohup [命令与参数] & --在终端机后台工作
但是这个命令需要注意的是,nohup并不支持bash的内置命令,所
- BusinessObjects Enterprise Java SDK
greemranqq
javaBOSAPCrystal Reports
最近项目用到oracle_ADF 从SAP/BO 上调用 水晶报表,资料比较少,我做一个简单的分享,给和我一样的新手 提供更多的便利。
首先,我是尝试用JAVA JSP 去访问的。
官方API:http://devlibrary.businessobjects.com/BusinessObjectsxi/en/en/BOE_SDK/boesdk_ja
- 系统负载剧变下的管控策略
iamzhongyong
高并发
假如目前的系统有100台机器,能够支撑每天1亿的点击量(这个就简单比喻一下),然后系统流量剧变了要,我如何应对,系统有那些策略可以处理,这里总结了一下之前的一些做法。
1、水平扩展
这个最容易理解,加机器,这样的话对于系统刚刚开始的伸缩性设计要求比较高,能够非常灵活的添加机器,来应对流量的变化。
2、系统分组
假如系统服务的业务不同,有优先级高的,有优先级低的,那就让不同的业务调用提前分组
- BitTorrent DHT 协议中文翻译
justjavac
bit
前言
做了一个磁力链接和BT种子的搜索引擎 {Magnet & Torrent},因此把 DHT 协议重新看了一遍。
BEP: 5Title: DHT ProtocolVersion: 3dec52cb3ae103ce22358e3894b31cad47a6f22bLast-Modified: Tue Apr 2 16:51:45 2013 -070
- Ubuntu下Java环境的搭建
macroli
java工作ubuntu
配置命令:
$sudo apt-get install ubuntu-restricted-extras
再运行如下命令:
$sudo apt-get install sun-java6-jdk
待安装完毕后选择默认Java.
$sudo update- alternatives --config java
安装过程提示选择,输入“2”即可,然后按回车键确定。
- js字符串转日期(兼容IE所有版本)
qiaolevip
TODateStringIE
/**
* 字符串转时间(yyyy-MM-dd HH:mm:ss)
* result (分钟)
*/
stringToDate : function(fDate){
var fullDate = fDate.split(" ")[0].split("-");
var fullTime = fDate.split("
- 【数据挖掘学习】关联规则算法Apriori的学习与SQL简单实现购物篮分析
superlxw1234
sql数据挖掘关联规则
关联规则挖掘用于寻找给定数据集中项之间的有趣的关联或相关关系。
关联规则揭示了数据项间的未知的依赖关系,根据所挖掘的关联关系,可以从一个数据对象的信息来推断另一个数据对象的信息。
例如购物篮分析。牛奶 ⇒ 面包 [支持度:3%,置信度:40%] 支持度3%:意味3%顾客同时购买牛奶和面包。 置信度40%:意味购买牛奶的顾客40%也购买面包。 规则的支持度和置信度是两个规则兴
- Spring 5.0 的系统需求,期待你的反馈
wiselyman
spring
Spring 5.0将在2016年发布。Spring5.0将支持JDK 9。
Spring 5.0的特性计划还在工作中,请保持关注,所以作者希望从使用者得到关于Spring 5.0系统需求方面的反馈。