HyperAttentionDTI: improving drug–protein interaction prediction by sequence-based deep learning wit
动机:识别药物-靶点相互作用(DTI)是药物再利用和药物发现的关键步骤。通过模型计算准确识别 DTI 可以显著缩短开发时间并降低成本。最近,提出许多基于序列的方法用于 DTI 预测,并通过引入注意机制来提高性能。这些方法仅对药物和蛋白质间的单个非共价间的分子相互作用进行建模,忽略了原子和氨基酸间复杂的相互作用。
结果:在本文中,我们提出了一种基于卷积神经网络和注意机制的端到端生物启发模型 HyperAttentionDTI。我们使用深度CNN来学习药物和蛋白质的特征矩阵。为了对原子和氨基酸间复杂的非共价分子间相互作用进行建模,我们利用特征矩阵上的注意机制,并为每个原子或氨基酸分配一个注意向量。我们在三个基准数据集上对 HpyerAttentionDTI 进行了评估,与最先进的基线相比,我们的模型实现了显著的性能。此外,对人类γ-氨基丁酸受体的个案研究证实,我们的模型可以用作预测DTI的有力工具。
1. Introduction
DTI预测

DTA预测

上述方法试图探索更强的模块来提取药物或蛋白质特征,但忽略了一个重要事实,即只有蛋白质的某些部分或药物的几个原子参与分子间相互作用,而不是整个结构。
为了对氨基酸和原子间的分子间相互作用建模,DTI 和 DTA 预测中引入了注意机制(Bahdanau等人,2015)。Tsubaki等人(2019)提出了一种基于注意力的 DTI 预测模型。该模型将药物编码为固定长度的载体,并使用单侧注意机制来计算蛋白质中的哪些子序列对分子更重要。Chen等人(2021)和Wang等人(2020)提出的模型也使用了这种注意机制。此外,Gao等人(2018)在 DTI 预测中应用了双侧注意机制以使药物和蛋白质能够相互感知。双侧注意机制不仅可以定位蛋白质上的结合位点,还可以探索原子在药物上的重要性。受Transformer(V aswani等人,2017)捕捉两个序列之间特征的强大能力的启发,Chen等人(2020)将药物和蛋白质视为两种序列,并提出了基于Transformer的模型 TransformerCPI。Huang等人(2021)还提出了一种基于Transformer的模型MolTrans,该模型在特征提取过程中引入Transformer编码器以捕捉药物或蛋白质子结构之间的语义关系。这些方法结合了注意机制来对药物和蛋白质间的单一的非共价分子间相互作用进行建模,并且比没有注意机制的模型获得更好的性能。但是,他们忽略了药物和蛋白质之间存在几种非共价相互作用类型的事实(例如氢键和π堆积)。
受之前基于注意力的模型的启发,我们提出了一种生物启发的端到端方法 HPyeratentionDTI。我们模型的输入是药物的SMILES序列和蛋白质的氨基酸序列,使用堆叠的1D-CNN层来从输入中学习特征矩阵。与以前基于注意力的模型不同,我们的模型为每个氨基酸原子对推断出一个注意力向量。这些注意向量不仅表示氨基酸和原子间的相互作用,而且控制通道上的特征表示。在注意力模块后,将修改过的药物-蛋白质特征向量输入到全连接网络中。在四种不同的药物发现环境下,我们将我们的模型与三种广泛使用的数据集上最先进的深度学习基线进行了比较。
2. Materials and Methods
2.1 Benchmark Datasets
我们从 DrugBank 数据库中提取药物和靶点数据以建立实验数据集。本研究中使用的数据于2020年1月3日发布(版本5.1.5)。我们手动丢弃无机化合物、小分子化合物(例如铁(DB01592)和锌(DB01693))或 RDKit python 包无法识别 SMILES 字符串的药物,最后,总共获得6655种药物、4294种蛋白质和17511种正DTI。按照惯例,我们从未标记的药物-蛋白质对中采样生成负样本,并获得具有相同正和负样本的平衡数据集。此外,我们还构建了两个不平衡基准数据集 Davis 和 KIBA。Davis 和 KIBA 记录了测量药物和蛋白质间结合亲和力的生物实验值。根据早期工作,分别为 Davis 和 KIBA 数据集设置阈值5.0和12.1,以构建二分类数据集。因为在 Davis 数据集中有一些具有相同氨基酸序列的蛋白质,我们删除了重复的药物-蛋白质对以避免标签混淆。
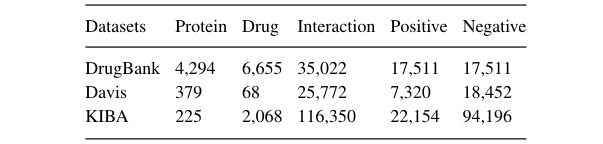
2.2 Proposed model
HyperAttentionDTI 由三部分组成:CNN模块、注意模块和输出模块。
给定药物的SMILES字符串和蛋白质的氨基酸序列,CNN模块从药物和蛋白质序列中提取特征矩阵,然后将特征矩阵输入到注意模块中以获得决策向量,输出模块根据决策向量进行预测。
2.2.1 Embedding layer
蛋白质的氨基酸序列和药物的SMILES序列是HyperAttentionDTI的输入。SMILES字符串由64个不同的字符组成,蛋白质中有20种不同的氨基酸。
HyperAttentionDTI从两个嵌入层开始,将每个氨基酸和SMILES字符转换为相应的嵌入向量,得到蛋白质嵌入矩阵 P e ∈ R M × e p P_e\in R^{M\times ep} Pe∈RM×ep 和 药物嵌入矩阵 D e ∈ R N × e d D_e\in R^{N\times ed} De∈RN×ed,其中 M M M 和 N N N 分别是蛋白质字符串和SMILES字符串的长度, e p ep ep 和 e d ed ed 分别是蛋白质字符串嵌入和SMILES字符串嵌入的大小。
2.2.2 CNN block
在我们的模型中有两个独立的 CNN 块,一个用于药物,另一个用于蛋白质。CNN 块包含三个连续的 1D-CNN 层,可以有效地提取序列语义信息。1D-CNN 能够从整个空间捕捉重要的局部模式。当卷积核在蛋白质或SMILES串上滑动时,可以捕获不同的氨基酸的组合或药物的子结构,并获得包含它们之间化学关系的潜在特征向量。给定嵌入层的嵌入矩阵 P e P_e Pe 和 D e D_e De,CNN块生成蛋白质和药物的潜在特征矩阵 P c n n ∈ R M × f P_{cnn}\in R^{M\times f} Pcnn∈RM×f 和 D c n n ∈ R N × f D_{cnn}\in R^{N\times f} Dcnn∈RN×f,其中 f f f 是最后一个1D-CNN层的卷积核的数量。
2.2.3 Attention block
HyperAttention 不仅在空间维度上对语义相关性进行建模,而且在药物子序列和蛋白质子序列间的通道维度上建模。给定药物蛋白质的潜在特征矩阵 D c n n D_{cnn} Dcnn 和 P c n n = { p 1 , p 2 , ⋯ , p M } P_{cnn}=\{p_1,p_2,\cdots,p_M\} Pcnn={p1,p2,⋯,pM},我们生成一个注意矩阵 A ∈ R N × M × f A\in R^{N×M×f} A∈RN×M×f,表示药物和蛋白质在空间和通道维度上的相互作用。
更准确地说,给定 d i d_i di 和 p j p_j pj,为了分离特征提取器和注意力模块,我们首先通过多层感知器(MLP)将它们转换为注意力向量 d a i da_i dai 和 p a j pa_j paj。 d a i = F ( W d ⋅ d i + b ) (1) da_i=F(W_d·d_i+b)\tag{1} dai=F(Wd⋅di+b)(1) p a j = F ( W p ⋅ p j + b ) (2) pa_j=F(W_p·p_j+b)\tag{2} paj=F(Wp⋅pj+b)(2)其中 F F F 是非线性激活函数(如: R e L U ReLU ReLU), W d ∈ R f × f W_d\in R^{f\times f} Wd∈Rf×f 和 W p ∈ R f × f W_p\in R^{f\times f} Wp∈Rf×f 是权重矩阵, b b b 是偏置向量。
注意向量 A i , j ∈ R f A_{i,j}\in R^f Ai,j∈Rf 计算方式如下: A i , j = F ( W a ⋅ ( d a i + p a j ) + b ) (3) A_{i,j}=F(W_a·(da_i+pa_j)+b)\tag{3} Ai,j=F(Wa⋅(dai+paj)+b)(3)其中 W a ∈ R 2 f × f W_a\in R^{2f\times f} Wa∈R2f×f 是权重矩阵。
经过上述操作后,我们得到注意矩阵 A ∈ R N × M × f A\in R^{N×M×f} A∈RN×M×f。通过对不同维度计算平均值,生成药物和蛋白质的注意矩阵 A d ∈ R N × f A_d\in R^{N×f} Ad∈RN×f 和 A p ∈ R M × f A_p\in R^{M×f} Ap∈RM×f: A d = Sigmoid ( M E A N ( A , 2 ) ) (4) A_d=\text{Sigmoid}\big(MEAN(A,2)\big)\tag{4} Ad=Sigmoid(MEAN(A,2))(4) A p = Sigmoid ( M E A N ( A , 1 ) ) (5) A_p=\text{Sigmoid}\big(MEAN(A,1)\big)\tag{5} Ap=Sigmoid(MEAN(A,1))(5)其中 M E A N ( I n p u t , d i m ) MEAN(Input,dim) MEAN(Input,dim) 是返回给定维度 d i m dim dim 中 I n p u t Input Input 的每行的平均值的平均运算, Sigmoid \text{Sigmoid} Sigmoid 是将所有注意力得分映射到 ( 0 , 1 ) (0,1) (0,1) 的激活函数。潜在特征矩阵 D a D_a Da 和 P a P_a Pa 更新公式如下: D a = D c n n ⋅ 0.5 + D c n n ⊙ A d (6) D_a=D_{cnn}·0.5+D_{cnn}⊙A_d\tag{6} Da=Dcnn⋅0.5+Dcnn⊙Ad(6) P a = P c n n ⋅ 0.5 + P c n n ⊙ A p (7) P_a=P_{cnn}·0.5+P_{cnn}⊙A_p\tag{7} Pa=Pcnn⋅0.5+Pcnn⊙Ap(7)其中 ⊙ ⊙ ⊙ 表示元素间乘积。
然后,我们对 D a D_a Da 和 P a P_a Pa 采用了 M a x P o o l i n g Max\ Pooling Max Pooling 操作来得到特征向量 v d r u g v_{drug} vdrug 和 v p r o t e i n v_{protein} vprotein。
2.2.4 Output block
输出模块由多层全连接网络(FCNN)组成。FCNN 的激活函数是负斜率为0.01的 L e a k y R e L U Leaky\ ReLU Leaky ReLU。每个 FCNN 后面都有一个 D r o p o u t Dropout Dropout 防止过拟合。最后一层输出相互作用可能性的概率 y ^ \hat{y} y^。作为二分类任务,我们使用二进制交叉熵损失来训练我们的模型: l o s s = − [ y log ( y ^ ) + ( 1 − y ) log ( 1 − y ^ ) ] (8) loss=-[y\log(\hat{y})+(1-y)\log(1-\hat{y})]\tag{8} loss=−[ylog(y^)+(1−y)log(1−y^)](8)其中 y y y 是标签。
2.3 Implementation
HyperAttentionDTI 在 PyTorch 中实现。对于优化参数,我们使用 AdamW 优化器,lr=0.0001,权重衰减系数为 0.0001,输入嵌入大小为64,这意味着我们用64维密集向量表示SMILES或氨基酸序列中的每个字符。每个CNN块由三个堆叠的 1D-CNN 层组成,分别具有32、64和96个卷积核。CNN块的窗口大小分别为 4、6、8(对于药物)和 4、6、12(对于蛋白质)。输出块由四个全连接层组成,其中神经元的数量分别为1024、1024、512和2,Dropout为0.1。我们将 batch size 设为 32。我们执行 early stopping 以解决过拟合问题。如果验证集上的模型丢失在20个时期内没有减少,则训练将停止。
3. Experiments and Results
3.1 Experiments setup
(1)Evaluation Strategies
假设 P t r a i n P_{train} Ptrain 和 D t r a i n D_{train} Dtrain 是训练集中的蛋白质和药物集合。当预测测试集中药物 d d d 和 蛋白质 p p p 之间的相互作用时,有如下四种不同的实验设置来进行综合比较:
- E 1 E_1 E1: d ∈ D t r a i n d\in D_{train} d∈Dtrain, p ∈ P t r a i n p \in P_{train} p∈Ptrain
- E 2 E_2 E2: d ∉ D t r a i n d\notin D_{train} d∈/Dtrain, p ∈ P t r a i n p \in P_{train} p∈Ptrain
- E 3 E_3 E3: d ∈ D t r a i n d\in D_{train} d∈Dtrain, p ∉ P t r a i n p \notin P_{train} p∈/Ptrain
- E 4 E_4 E4: d ∉ D t r a i n d\notin D_{train} d∈/Dtrain, p ∉ P t r a i n p \notin P_{train} p∈/Ptrain
我们进行了10次重复5折交叉验证来评估模型的预测能力。对于每一次,我们在不同的随机种子下对数据集进行不同的随机分割。
(2)The search of hyper-parameters
在我们的模型中有四个重要的超参数,即学习率、权重衰减系数、批量大小和 dropout。这些超参数由DrugBank数据集上的网格搜索确定。在网格搜索中,学习率在[1e-1、1e-2、1e-3、1e-4、1e-5、1e-6、1e-7]中,批次大小在[8、16、32、64、128、256、512]中,权重衰减系数在[1e-2、1-e-3、1-e-4、1-e-5、1-e-6、1-e-7],辍学率在[0.1、0.2、0.3、0.4、0.5]中。一般来说,学习率直接决定性能,批次大小与学习率相关。因此,我们首先在网格搜索中确定学习率和批量大小。在学习率和批量大小固定后,我们选择权重衰减系数和 dropout 来提高模型的鲁棒性。优化的学习率、权重衰减系数、批次大小和辍学率分别为 1e-4、1e-4、32 和 0.1。
3.2 Baselines
- GNN-CPI(2019):通过图神经网络和1D-CNN对药物和蛋白质进行编码,并使用单侧注意机制来考虑蛋白质中的子序列对药物的重要性,并且将药物和蛋白质的特征向量连接并送入 FCNN 以预测 DTI。
- GNN-PT(2020):用 GNN 提取药物特征向量,Transformer 和 CNN 提取蛋白质表征,使用单侧注意机制来获得蛋白质特征向量,然后将其与药物特征向量连接并送入 FCNN 进行最终预测。
- DeepEmbedding-DTI(2021):分别通过 GNN 和 BiLSTM 对药物和蛋白质编码,并使用基于 Transformer 的模型学习蛋白质序列的嵌入向量。
- GraphDTA(2020):分别使用 GNN 和 CNN 提取药物和蛋白质特征,选择 GAT_GCN 作为特征提取器,并在最后一层之后添加了一个 Sigmoid \text{Sigmoid} Sigmoid 函数将其改为 DTI 预测。
- DeepConv-DTI(2019):用 FCNN 处理药物的 ECFP 指纹,并应用多尺度1D-CNN和全局最大池化层提取蛋白质序列中的各种长度的局部模式。然后,将药物和蛋白质的抽象特征向量连接并送入 FCNN 以预测 DTI。
- TransformerCPI(2020):基于 Transformer 架构,将药物和蛋白质视为两种序列。分别从 CNN 和 GCN 生成蛋白质序列特征和原子特征后,TransformerCPI 通过 Transformer 的解码器获得相互作用特征,并使用线性层输出相互作用的概率。
- MolTrans(2021):使用频繁的连续子序列挖掘模块将药物和蛋白质分解为一组明确的子结构序列,然后利用 Transformer 嵌入模块获得药物和蛋白质的增强上下文嵌入。接下来,MolTrans 通过点积对相互作用图进行建模,并在相互作用图上应用 CNN 和 FCNN 来预测 DTI。
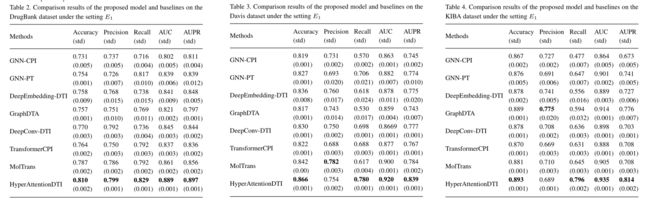
3.3 Performance evaluation under the setting E 1 E_1 E1
首先在设置E1下将我们的模型与DrugBank数据集上的基线进行比较。我们以16:4:5的比例将DrugBank数据集划分为训练、验证和测试集。
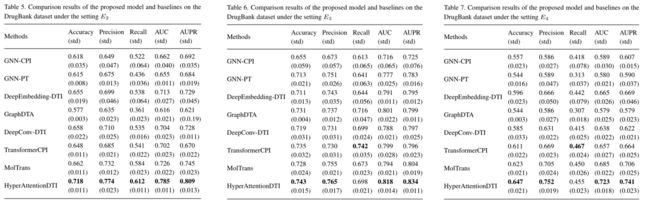
3.4 Performance evaluation under de novo setting
为了测试我们模型的健壮性,我们在 DrugBank 数据集上的设置E2、E3和E4下评估我们的模型和基线。在药物发现方面,设置E2和E3比设置E1更真实。为了在E2/E3设置下测试这些模型,我们随机选择20%的药物/蛋白质,并将所有与这些药物/蛋白质相关的DTI作为测试集。其他DTI用作训练集和验证集,比率为4:1。
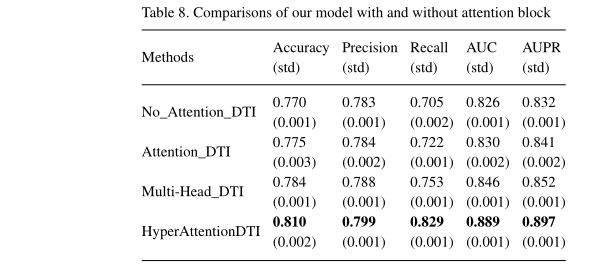
3.5 The effectiveness of Attention block
为了评估注意块的重要性,我们提出了三个子模型。
- 无注意力机制:直接对CNN块的输出应用全局最大池化运算,以获得药物和蛋白质特征向量,连接向量并将其输入输出块以进行预测。
- 基于双向注意力机制:给定药物特征矩阵 D c n n D_{cnn} Dcnn 和蛋白质特征矩阵 P c n n P_{cnn} Pcnn,注意力权重计算方式如下: A i = Sigmoid ( D c n n ⋅ P c n n T ) (9) A_i=\text{Sigmoid}(D_{cnn}·P_{cnn}^T)\tag{9} Ai=Sigmoid(Dcnn⋅PcnnT)(9)
- 基于多头注意力机制:对于每个头 i i i,注意力权重计算方式如下: D a i = F ( W d i ⋅ D c n n + b ) (10) Da_i=F(W_{d_i}·D_{cnn}+b)\tag{10} Dai=F(Wdi⋅Dcnn+b)(10) P a i = F ( W p i ⋅ P c n n + b ) (11) Pa_i=F(W_{p_i}·P_{cnn}+b)\tag{11} Pai=F(Wpi⋅Pcnn+b)(11) A i = Sigmoid ( D a i ⋅ P a i T ) (12) A_i=\text{Sigmoid}(Da_{i}·Pa_{i}^T)\tag{12} Ai=Sigmoid(Dai⋅PaiT)(12)其中 W d i ∈ R f × d W_{d_i}\in R^{f\times d} Wdi∈Rf×d 和 W p i ∈ R f × d W_{p_i}\in R^{f\times d} Wpi∈Rf×d 是权重矩阵。最终注意矩阵 A ∈ R N × M × f A\in R^{N\times M\times f} A∈RN×M×f计算方式如下: A = 1 K ⋅ ∑ i = 0 K A i (13) A=\frac{1}{K}·\sum_{i=0}^KA_i\tag{13} A=K1⋅i=0∑KAi(13)其中 K K K 是头的数量,多头注意力机制还具有模拟原子和氨基酸之间复杂相互作用的能力,然而,这种机制引入了大量的模型参数,其数量取决于超参数K。通常设 K = 8 K=8 K=8。
值得一提的是,我们在注意力块中尝试了不同的激活功能,发现ReLU功能取得了最佳效果。我们推测这与提取的药物和蛋白质特征矩阵有关。
3.6 Case studies
为了评估我们模型的可靠性,我们使用 FDA 批准的靶向特定蛋白质的药物 人γ-氨基丁酸受体(GABAR)进行了一项案例研究。选择 GABAR 进行案例研究,因为它们是中枢神经系统中最重要的抑制性氯离子通道,是多种药物的主要靶点。GABAR中有7个亚基和16个靶蛋白。我们计算了16种 GABAR 蛋白和6708种药物之间的相互作用概率,并根据它们的概率进行排序。下表描述了在训练和测试集中划分的药物数量,最后一列显示了前10名列表中预测的药物数量。

3.7 Model interpretation
为了证明注意机制不仅提高了模型的性能,而且导致了更多的可解释性,我们进行了两个案例研究,即与GW0385(PDB:2FDD)结合的HIV蛋白酶D545701的晶体结构和无乳链球菌与抑制剂AT018(PDB:5JF3)复合的2型PDF的晶体结构。我们首先将药物SMILES和氨基酸序列输入我们的模型,然后得到蛋白质注意力矩阵 A p ∈ R M × f A_p\in R^{M\times f} Ap∈RM×f 我们将平均算子应用于 A p A_p Ap 以获得蛋白质注意向量 a p ∈ R M a_p\in R^M ap∈RM(反映了氨基酸序列上注意力值的分布)。然后,我们将注意向量 a p a_p ap 映射到复合物的3D结构,以可视化蛋白质中哪些区域对相互作用具有更有效的作用。
2FDD和5JF3的注意力权重如图所示。蛋白质中获得高注意力权重的氨基酸在3D结构可视化中以红色突出显示。如图a所示,12个结合位点中的两个,ALA 28和PRO 81获得了高关注度得分,尤其是PRO 81得分最高。对于5HF3,有10个结合位点。如图b所示,LEU 132获得第四高分,VAL 71获得最高分。
这些结果表明,我们的模型可以帮助研究人员缩小结合位点的搜索空间。同时,我们也注意到许多非结合位点被突出显示。
