
Elaine Mardis教授的介绍可能有点略长,那我就在开头做一个小结好了,就算读不下去了也能知道个大概:
Elaine Mardis教授,俄亥俄州哥伦布市全国儿童医院(Nationwide Children’s Hospital, Columbus, OH, USA),基因组医学研究所联合执行主任,现任美国癌症研究学会(AACR)主席,这里主要介绍与Elaine Mardis教授有关的五件影响力大的事情:
- 2018年《Nature》邀请了Elaine Mardis教授等6位业内权威展望了2018年生命科学行业发展的技术趋势。她对开发癌症疫苗做出来自己的见解。
- 与来自西班牙和澳大利亚的科学家一起发现旨在靶向特殊基因突变的抗癌药物的疗效依赖于涉及的肿瘤组织的类型以及基因突变的性质。这项研究发表于《Nature》的新研究中。
- 另一篇乳腺癌肿瘤异质性相关的文章发表在《 Nature Communications》上,这项研究表明,降低ER阳性乳腺癌的雌激素水平改变了肿瘤的遗传学,这些改变对于指导手术切除肿瘤后患者的治疗非常重要。
- 《Nature》旗下《British Journal of Cancer》 (BJC)上发表研究报告,通过对乳腺早期病变组织的分子学分析,探讨了乳腺癌的分子学发病机制。
- 在 Frontiers in Cancer Research, October 2018, Heidelberg, Germany 会议上做主讲人。本次报告主要针对儿童癌症病例中的复发或难治病例,应用基因组学技术和先进的分析方法进行研究和诊断其中遇到的困难,现行方法,成功和失败的经验进行总结,并对未来的趋势进行展望。
1.Nature:2018年技术展望之开发癌症疫苗
在癌症的免疫基因组学领域,研究者们想知道在某个特定的人身上,哪个癌症基因组编码的蛋白的突变会引起免疫应答。这类蛋白称为“新抗原(neo-antigens)”,可利用来研究个体化的癌症疫苗,或提示其他治疗方法。
有种令人振奋的技术可用于研究这类新抗原,叫CyTOF,质谱流式细胞术,用来鉴定表达某种特定蛋白的细胞。
在典型的流式细胞术中,研究者们将标记有荧光分子的抗体与细胞混合,从而标记感兴趣的蛋白。然后细胞们一个个被分析,测定那些蛋白的相对丰度。但其局限性在于荧光标签屈指可数,而CyTOF则采用金属标签,在质谱仪中检测——一次可检测上百种不同的标签,而同样条件下流式细胞可能只有十来个标签。
这项技术可能会改变癌症免疫基因组学领域,让研究者们能找到一个人的癌细胞中,哪种新抗原的丰度最高,最可能激起免疫系统的强烈反应。于是研究者便可利用这些信息来制造个体化的抗癌症“疫苗”。新的免疫检查点抑制剂联合应用,可让癌症患者自己战胜病魔。
但对于任何由基因组预测到的新抗原,它是否真的会起显著的免疫反应仍是个推测性的工作。CyTOF则让我们得以定量检测多种预测的多肽与个人T细胞的结合强度,从而进一步确认其可能性。
此外,它也并不仅仅是用于癌症基因组学研究。CyTOF可用来检测细胞中各种蛋白的丰度,只要你能找到抗体来跟你的目标蛋白结合。它让我们得以从蛋白质层面讨论问题,比过去的方法维度更多,精度更高。
2. 《Nature》文章介绍
文章:David M. Hyman et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers, Nature (2018). DOI: 10.1038/nature25475
过去医生都以相同的方式治疗同一种肿瘤病人,但是最近的研究发现这可能不是最佳的策略。这是因为不是所有同一类的肿瘤都相似。比如不是所有的肺癌病人的肿瘤都一样——每个病人的诱因和发展都不一样。考虑到这个因素,科学家们已经在寻找方法基于病人自身情况给出个性化治疗方案。由于这种方法已经取得了一定的进展,科学家们也发现肿瘤的大部分不同都可以归结于基因不同。在这项新研究中,研究人员聚焦于ERBB2和ERBB3基因,因为已有研究发现它们可以过表达HER2蛋白。
为了研究ERBB2和ERBB3基因在肿瘤发展中发挥的作用,研究人员在临床实验中使用来那替尼治疗了141个病人(共患有21种癌症,42种突变),它可以抑制HER的产生。他们发现这个药物的效果依赖于突变和组织环境。一些病人携带ERBB2突变,他们对这个药物响应很好,尤其是那些HER2胞外段或者是激酶结构域出现突变的病人。而其他表达HER2的病人对该药物响应不太好,而携带ERBB3的病人对这个药物根本就不产生反应。
研究人员认为他们的研究表明针对特定突变的疗法必须考虑肿瘤组织以及肿瘤组织涉及的组织性质。
3. 乳腺癌肿瘤异质性文章—《 Nature Communications》
文章:Aromatase inhibition remodels the clonal architecture of estrogen-receptor-positive breast cancers. Nature Communications, doi:10.1038/ncomms12498
该研究表明,雌激素受体(ER)阳性乳腺癌患者接受激素减量治疗后表现出克隆复杂性,因此临床上应该对患者治疗前后的肿瘤基因组进行比较分析,以便得到指导治疗的重要信息。
来自华盛顿大学医学院麦克唐奈基因组研究所(McDonnell Genome Institute)和贝勒医学院(Baylor College of Medicine)的研究人员对原发性肿瘤样本进行了全基因组测序,这些样本来自于22名患有II 期或 III期乳腺癌的绝经后妇女,取用了她们在接受芳香化酶抑制剂新辅助激素治疗前后的肿瘤样本。研究小组还对相匹配的正常样本进行了基因组测序,对肿瘤样本进行了RNA测序。
通过分析这些数据,研究人员发现在治疗前后以及肿瘤的不同部分存在突变模式和克隆群体的显著异质性。结合治疗效果分析发现,雌激素受体阳性乳腺癌具有遗传多样性,并且在激素治疗后出现大量突变。
文章共同通讯作者Matthew Ellis说,“在治疗造成的环境压力下,肿瘤会形成新的亚克隆,这些克隆会在治疗环境下生存并生长,这也就是为什么我们会在治疗ER阳性乳腺癌的最后阶段出现困难。我们在大部分肿瘤中都发现了这个情况。”
研究人员重点关注了22名绝经后女性患者,她们患有ER阳性luminal A型或luminal B型乳腺癌,其中12个人患有芳香化酶抑制剂敏感型肿瘤,另外10个出现芳香化酶抑制剂抗性。
来自这些患者的肿瘤样本曾在一项试点研究中被测序过,研究小组又对手术切除样本进行了基因组测序,以了解肿瘤在接受芳香化酶抑制剂新辅助治疗前和接受治疗四个月后出现的遗传模式。
治疗前后肿瘤突变模式差异显著
研究人员还对20个基线时的肿瘤样本和18个手术时采集的肿瘤样本进行了RNA测序,另外还对其他几十个病例的肿瘤样本进行了靶向基因panel测序。
结果发现,在这22名乳腺癌患者中,2名患者治疗前后的肿瘤突变模式保持相对稳定,其余20名患者则出现了更加复杂的突变模式。
在其中2个病例中,研究小组揭示了所谓“碰撞瘤”在遗传上是完全不同的,碰撞瘤是指同一个体中两个独立的原发性肿瘤相互碰撞或浸润而形成的肿瘤。
根据RNA测序和肿瘤免疫组化数据,治疗前被分类为ER阳性的乳腺肿瘤,在接受芳香化酶抑制剂新辅助治疗后肿瘤中同时出现ER阳性和ER阴性。
在另外18个病例中,肿瘤在治疗前后出现了明显不同的肿瘤突变模式和复杂的克隆转变。例如在一个患者中,基线时在大部分细胞中检测到的亚克隆,在治疗后几乎被基线时只存在于2%细胞中的另一个亚克隆替代。
另外57个乳腺癌病例的靶向测序和全基因组测序数据也表现出类似的复杂性。研究小组在治疗后的肿瘤序列中发现了抗性突变猛增,包括ESR1基因变异。
文章共同通讯作者Elaine Mardis说,“这项研究表明,降低ER阳性乳腺癌的雌激素水平改变了肿瘤的遗传学,这些改变对于指导手术切除肿瘤后患者的治疗非常重要。”
她表示,在手术前几个月接受芳香化酶抑制剂治疗的患者在手术前应该及时进行重新评估,以确定肿瘤响应治疗而发生的改变。这种信息可以帮助我们预测,进一步的雌激素抑制治疗是否有助于降低复发风险。
4. 乳腺癌的分子学发病机制研究—《British Journal of Cancer》

文章:Somatic mutations in benign breast disease tissue and risk of subsequent invasive breast cancer.
2018年6月6日,英国癌症研究基金会和英国《自然》旗下《英国癌症杂志》在线发表 美国纽约爱因斯坦医学院、圣路易斯华盛顿大学麦克唐奈基因组研究所、布朗大学阿尔伯特医学院罗德岛医院、哈肯萨克大学医学中心、凯泽永久医疗集团医疗研究中心、俄亥俄州立大学医学院全国儿童医院基因组医学研究所的研究报告,通过对乳腺早期病变组织的分子学分析,探讨了乳腺癌的分子学发病机制。
该病例对照研究数据来自凯泽永久医疗集团西北地区1971~2006年经活检确认被诊断为良性乳腺病变并随访至2015年中期以确定随后是否发生浸润性乳腺癌的女性队列;其中,病例组为良性乳腺病变随后发生浸润性乳腺癌女性218例,对照组为与病例组按1∶1配对的良性乳腺病变相同随访间期未发生浸润性乳腺癌女性。对83个乳腺癌重要基因进行目标序列捕获并测序。
结果发现,病例组与对照组相比,突变总负荷、非沉默突变、个别基因、基因突变性质、通路水平突变富集无显著差异。来自7例受试者良性乳腺病变与同侧浸润性乳腺癌的DNA几乎没有相同突变。
因此,该研究首次通过靶向多基因测序法,调查了早期乳腺癌前病变的全部基因,结果表明良性乳腺病变组织检测到的体细胞基因突变与乳腺癌风险无关。
5. Frontiers in Cancer Research 会议
基于亚马逊云服务的基因组分析(AWS-based genome analysis)
目前Nationwide Children’s Hospital的分子诊断平台(GenomeNext’s Platform)使用Amazon Web Service(AWS)云服务器,可每天分析1000个基因组。其中80%的病人样本来自于冰冻组织(fresh frozen samples),20%的病人样本来源于福尔马林固定的石蜡包埋组织(FFPE samples)。在病人签署同意书之后进行全基因组(WGS, 深度60x)、全外显子组(WES, 深度250x)的肿瘤和对照正常组织测序和肿瘤组织的RNA测序(RNAseq); 还有一部分病例在病患孩子家长同意之后会对家长或者其他家庭成员进行采血后测序。这种体系可以充分涵盖体细胞突变及可遗传突变(somatic mutations & germline mutations)。
二代测序(NGS)分析的临床应用
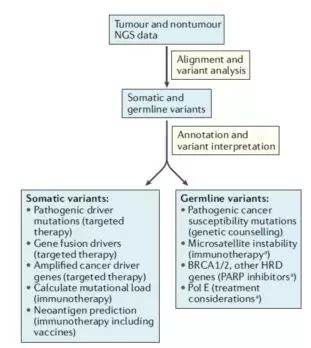
通过肿瘤细胞和正常体细胞的对比,建立变异分析检测标准流程(图1)。和用药相关的体细胞突变分析主要包括有靶向药针对的致病驱动基因、融合基因、扩增基因,免疫疗法相关的突变量计算和新抗原预测。而可遗传突变主要针对与癌症相关性高的多态性位点(SNP),微卫星序列(microsatellite),和PARP抑制剂相关的突变,如:BRCA1/2, 同源DNA重组修复缺陷(HRD),DNA聚合酶ε (如:Pol E基因突变的患者肠癌复发的机率显着减少)。其他分析包括临床意义不明确的突变、扩增、删除、小片段插入以及缺失(Indel)、染色体结构变化、肿瘤内异质性(intratumoral heterogeneity)等等。
树屋儿童癌症公共数据(The Treehouse Childhood Cancer Initiative)
加州大学圣克鲁兹分校基因组学研究所(UCSC Genomics Institute)的树屋儿童癌症倡议使用共享数据分析儿童肿瘤和成人肿瘤(网站链接见后)。该数据库共收集了来自11000多个肿瘤样本的数据,并可供所有研究人员免费下载和进行可视化。 该基因表达数据与患者隐私保护的临床数据一起可用,包括年龄,性别和疾病类型等。最近发布的数据版本8 (发布于2018年7月)包含11427个poly-A 筛选-核糖体去除的RNA样本的RNAseq数据及临床数据,以器官系统排序,相同种类的肿瘤可以进行比较,结果包括差异基因表达,信号通路富集,基因融合等。其中融合基因全部由PCR二次确认,并包含表达量排序。
数据库中包含多个病理无法确诊或较难确诊的疑难、罕见病例,最终通过RNA-seq分子诊断找到致病分子机理,从而为病人选择了合适的个性化靶向治疗方案,如:
病人1患有FGFR1:TACC1融合基因的胶质母细胞瘤(glioblastoma) ,后应用FGFR抑制剂治疗;
病人2患有MET-RBPMSLP融合基因的梭形细胞肉瘤 (spindle cell sarcoma), 后应用MET抑制剂治疗;
病人3青少年复发性脑瘤患者(relapsed childhood CNS tumor),家族性遗传NF-1基因突变,基因组改变复杂,经过信号通路富集分析和癌症免疫微环境分析发现该病人巨噬细胞增多,PD1表达较其他glioblastoma病人增高,酪氨酸激酶表达量增高等,后决定将其纳入anti-PD1和MEK抑制剂联合应用的成人临床试验。
临床应用NGS的挑战
二代测序在临床上应用的挑战大体可以从测序技术本身、病人样本、数据分析和临床解释几个方面来说。随着测序数据的复杂化和方法的多样化,对测序结果的临床意义解释成为这个领域中最大的难点。一种癌症中有哪些驱动基因、相同突变在不同癌症中意义不同、什么样的变化可以算作可治疗的突变(targetable genomic alterations)等等很多问题仍然有待基础科研去解决。病人样本的采集方式和对应的分析方法有很多种,哪种既能准确地反映不同时期的肿瘤特性,又能简单易行。病人的样本通常是稀缺资源,数量非常有限,生物检材在日常处理中不容易保证样品处理的一致性,极易降解和污染,这些都会直接影响测序数据的结果。数据分析方面,由于不同种类的改变需要不同的算法来计算,检测的灵敏度会受到测序深度的影响,因此NGS分子诊断通常需要保证高深度测序和一定广度的覆盖范围。另外由于全基因组数据量巨大,分析时间往往较长,不足以满足临床快速诊断的需要,优化出高效且精准的标准分析流程成为必须。在当前条件下即便有很大的云计算服务器,全基因组(WGS)和全外显子组(WES)数据依然需要较长时间和大量人力去分析解读,不同平台和工作流程之间产生结果的可重复性仍需提高。还有大数据产出之后的储存和访问等等,都是分子诊断中的挑战。
数据和软件共享(Data and software sharing):
由于儿童癌症相对罕见,所以共享数据和临床资料对了解疾病十分重要。整合多平台数据,从聚焦单一驱动基因到信号通路,可以更好地了解每个病人的癌症基因组。建立更多方便下载的整合工具包和基于网络的数据库,增加专职基因数据分析人员和提高病人数据访问权限,将构成未来的主要工作内容。
结论:
虽然面临诸多挑战,针对儿童癌症的临床基因组学检验依然可以为治疗计划,诊断和预告提供重要的临床证据。
结合DNA和RNA的数据可以很有效地提供癌症驱动基因(driver genes),新抗原(neoantigens)和免疫微环境(immune microenvironment)的信息,但是需要评估各类治疗方案的临床获益(clinical benefit)和分子水平变化上的关系。
分享数据和软件,包括分享治疗信息和临床结果对儿童癌症来说至关重要,甚至只发表一小部分针对和治疗相关的分子特性的探索性研究都十分重要。
对患者群体的教育会促进这些诊断和检测方法的信息透明化, 但是需要更多创新方法和开源数据库的支持。