EMBOSS的安装以及使用
EMBOSS 是欧洲分子生物学开放软件包,主要做序列比对,数据库搜搜,蛋白模块 分析和功能域分析,序列模式搜索,
引物设计等。
一些常见的应用。具体使用及功能可以参照http://emboss.sourceforge.net/apps/
或者在/EMBOSS-6.6.0/emboss路径下,输入./prophet --h 查看(仅仅输入prophet --h 会提示The program 'prophet' is currently not installed. To run 'prophet' please ask your administrator to install the package 'emboss' )
prophet Gapped alignment for profiles.
infoseq Displays some simple information about sequences.
water Smith-Waterman local alignment.
pepstats Protein statistics.
showfeat Show features of a sequence.
palindrome Looks for inverted repeats in a nucleotide sequence.
eprimer3 Picks PCR primers and hybridization oligos.
profit Scan a sequence or database with a matrix or profile.
extractseq Extract regions from a sequence.
marscan Finds MAR/SAR sites in nucleic sequences.
tfscan Scans DNA sequences for transcription factors.
patmatmotifs Compares a protein sequence to the PROSITE motif database.
showdb Displays information on the currently available databases.
wossname Finds programs by keywords in their one-line documentation.
abiview Reads ABI file and display the trace.
tranalign Align nucleic coding regions given the aligned proteins.
源码安装EMBOSS, 下载地址ftp://emboss.open-bio.org/pub/EMBOSS/emboss-latest.tar.gz.
参照http://emboss.sourceforge.net/download/
1.解压到目录
2.执行命令 ./configure 生成Makefile文件(./configure --prefix=/home/ct/soft/specific_name(不在指定目录时添加))
3.make
4.make install(可无)
5.测试是否编译成功
进入安装目录下的emboss路径,将测试输入文件复制到自己定义的一个目录
# 仅仅是安装完emboss,(可以把bin文件添加到系统环境变量中)做引物方面的分析还是不行,还需要安装个primer3,链接:http://primer3.sourceforge.net/
安装步骤:/software$运行以下命令
url=https://sourceforge.net/projects/primer3/files/primer3/2.3.7/
wget ${url}primer3-2.3.7.tar.gz -O primer3-2.3.7.tar.gz
tar xvzf primer3-2.3.7.tar.gz
cd primer3-2.3.7/src
make all
#确保~/bin在环境变量中
ln -s `pwd`/primer3_core ~/bin/primer32_core(在bin中创建一个软链接)
以上步骤运行完成后,运行eprimer32 -sequence test.fa -outfile test.fa.primer \
-targetregion 0,371 -optsize 20 -numreturn 3 \
-minsize 15 -maxsize 25 \
-opttm 50 -mintm 45 -maxtm 55 \
-psizeopt 200 -prange 100-280
会提示targetregion 0,371有问题,由于目前没有找到原因,所以就运行最简单的命令,eprimer32 -sequence test.fa -outfile test.fa.primer
这时会提示Error: thermodynamic approach chosen, but path to thermodynamic parameters not specified错误。
解决方案:
一:输入命令时加入-default_version=1的参数(发现还是不行)
发现Primer3文档中有这样的改变:
“2.5. IMPORTANT: because PRIMER_THERMODYNAMIC_OLIGO_ALIGNMENT=1,PRIMER_THERMODYNAMIC_PARAMETERS_PATH must point to the right location.This tag specifies the path to the directory that contains all theparameter files used by the thermodynamic approach. In Linux, thereare two *default* locations that are tested if this tag is notdefined: ./primer3_config/ and /opt/primer3_config/. For Windows,there is only one default location: .primer3_config. If the theparameter files are not in one these locations, be sure to setPRIMER_THERMODYNAMIC_PARAMETERS_PATH.”
二 sudo mkdir /opt/primer3_config
sudo cp -R primer3-2.3.7/src/primer3_config/* /opt/primer3_config
以上所有操作完成后才是完整的。
测试:
测试数据(自己创建一个test.fa文件)
cat <
>comp24_c0_seq1
TTACTCTCATCCTCCCCTTGTTGAAAGATTGGCTGCAATTGATGAACCCGATAAGAAGGTCAACTAAGAGAAGTGTAC
TTTTACGCATGGCATGGCATGGCGAGATATGGCTGTAATATGAGTATTATTTTCCTATGTTGCTACCGATATTTTCTA
TTTGCATATGAAAATTCCAAACCCAGAGTTAGGGGCCATATCTAAAGGGAATTTGCTAACGAGTAAATGGGAAAATAG
GAAATGTCAGAGGAGAtagcctagcctagcctagcctagccTCGCCTCATGTAACGAAATACAATTTAAATTTTGCTT
TACAGCTAATAGTCAGACTTTACATTTTGCTAAAA
END
①设计引物
eprimer32 -sequence test.fa -outfile test.fa.primer
引物结果:
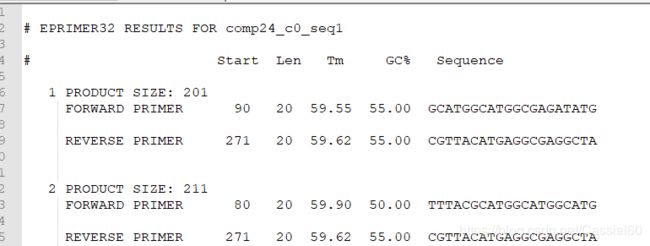
②整理引物格式位PrimerSearch 需要的格式
awk '{if($0~/EPRIMER32/) {seq_name=$5;count=1;} else \
if($0~/FORWARD PRIMER/) forward=$7; else if ($0~/REVERSE PRIMER/) \
{reverse=$7; printf("%s@%d\t%s\t%s\n", seq_name,count,forward, reverse); \
count+=1;} }' test.fa.primer >all_primer_file
结果:
comp24_c0_seq1@1 GCATGGCATGGCGAGATATG CGTTACATGAGGCGAGGCTA
comp24_c0_seq1@2 TTTACGCATGGCATGGCATG CGTTACATGAGGCGAGGCTA
comp24_c0_seq1@3 GCATGGCATGGCGAGATATG TTCGTTACATGAGGCGAGGC
comp24_c0_seq1@4 ATGGCATGGCGAGATATGGC CGTTACATGAGGCGAGGCTA
comp24_c0_seq1@5 GGCATGGCATGGCGAGATAT CGTTACATGAGGCGAGGCTA
③模拟PCR
primersearch -seqall test.fa -infile all_primer_file -mismatchpercent 5 -outfile test.database.primerSearch
结果:

needleall 的使用
needleall 读入两个文件,第一个文件的每个序列都与第二个文件的每个序列进行全局比对,采用Needleman-Wunsch
算法。
#随机生成测试数据
cat <
BEGIN{srand(seed); seq[0]="A"; seq[1]="C"; seq[2]="G"; seq[3]="T"}
{for(i=1;i<=chrNum;i++)
{print ">"label""i; len=(10-int(rand()*10)%2)/10*expected_len;
for(j=0;j<=len;j++) printf("%s", seq[int(rand()*10)%4]); print "";
}
}
END
echo 1 | awk -v seed=$RANDOM -v label=mm -v chrNum=2 -v expected_len=40 -f generateRandom.awk >test1.fa
echo 1 | awk -v seed=$RANDOM -v label=hs -v chrNum=2 -v expected_len=40 -f generateRandom.awk >test2.fa
needleall -asequence test1.fa -bsequence test2.fa -gapopen 10 -gapextend 0.5 -outfile test12.needle.alignment -auto -aformat3 pair
结果:
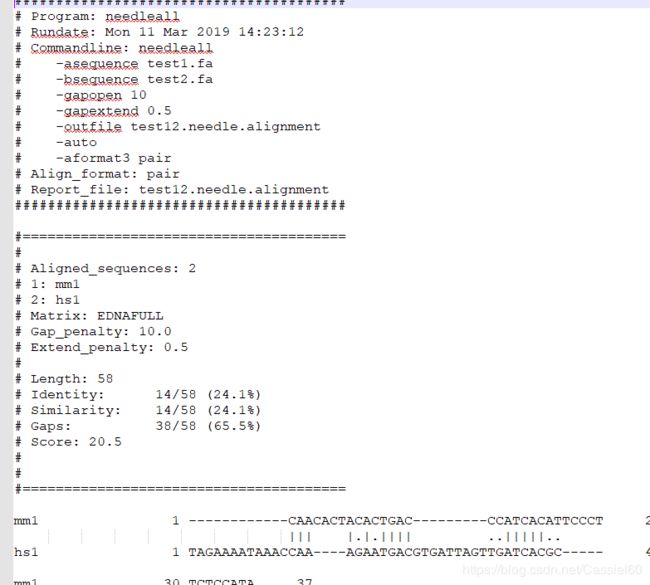
needleall -asequence test1.fa -bsequence test2.fa -gapopen 10 -gapextend 0.5 -outfile test12.needle.score -auto
结果:
mm1 hs1 58 (20.5)
mm2 hs1 57 (32.0)
mm1 hs2 49 (31.5)
mm2 hs2 47 (31.0)
在绝对路径下执行命令./needleall -asequence ../test.fa -bsequence ../test.fa -auto -aformat3 pair -sprotein1 1 -sprotein2 1 -outfile out.aln