RDKit|分子3D构象生成与优化
文章目录
- 一、构象生成算法概述
- 1.基于距离(distance-based)
- 2.基于知识(knowledge-based)
- 二、代码实现
- 1.添加氢原子
- 2.距离几何算法生成3D结构
- 3.距离几何+ETKDG生成3D构象
- 4.距离几何+ETKDG生成多构象
- 5.距离几何+ETKDG+MMFF生成3D构象
- 6.距离几何+ETKDG+MMFF生成多构象
- 7.多线程生成多构象
- 三、结语
一、构象生成算法概述
先来了解两种rdkit生成3D构象的方法:
1.基于距离(distance-based)
传统的距离几何(Distance Geometry)法生成构象步骤:
- (1) 通过分子的连接表信息和一套规则,生成分子的连接边界矩阵(molecule’s distance bounds matrix )
- (2) 使用三角形边界平滑算法(triangle-bounds smoothing algorithm),对边界矩阵进行平滑处理
- (3) 根据边界矩阵,随机产生一个距离矩阵。
- (4) 把产生的距离矩阵映射到三维空间中,并为每个原子计算坐标。
- (5) 对计算的坐标结果使用力场和边界矩阵进行粗略的优化。
这种方法虽然计算速度快,也能得到分子的三维坐标,但纯粹基于距离计算会导致部分结构的扭曲,也就是结果比较丑。想得到更好的结构,需要进行更细致的力场优化,比如再使用rdkit的通用力场(Universal Force Field,UFF)进行处理。
2.基于知识(knowledge-based)
Riniker和Landrum开发了一种(Experimental-Torsion Basic Knowledge Distance Geometry,ETKDG)的方法。他们从晶体结构数据库的小分子结构中总结了一些规则(扭转角的倾向性,torsion angle preference,符合某种SMARTS的结构更倾向于生成某种固定的构象),并用这套规则来修正距离几何算法产生的构象。如果使用了ETKDG,就可以不再使用力场进行优化了。
在rdkit中,目前已经将ETKDG作为默认的构象生成方法。使用代码实现这种方法比上面的废话都要简单,几行代码就能搞定了。
>>> from rdkit import Chem
>>> from rdkit.Chem import AllChem
>>> from rdkit.Chem import Draw
>>> m = Chem.MolFromSmiles('c1ccccc1')
>>> m3d=Chem.AddHs(m)
>>> AllChem.EmbedMolecule(m3d, randomSeed=1)
>>> Draw.MolToImage(m3d, size=(250,250))
二、代码实现
1.添加氢原子
- 加氢:Chem.AddHs()
在rdkit中,分子在默认情况下是不显示氢的,但氢原子对于真实的几何构象计算有很大的影响,所以在计算3D构象前,需要使用Chem.AddHs()方法加上氢原子。 - 减氢:Chem.RemoveHs()
相对应的,可以使用该方法把氢去除。
>>> m = Chem.MolFromSmiles('CC1=CC=C(C=C1)NC2=C3C(=C(C=C2)NC4=CC=C(C=C4)C)C(=O)C5=CC=CC=C5C3=O')
>>> m3d = Chem.AddHs(m)
>>> print(m.GetNumAtoms(), m3d.GetNumAtoms())
32 54
2.距离几何算法生成3D结构
使用距离几何算法初始化3D坐标。
- 生成3D构象:AllChem.EmbedMolecule(mol, randomSeed, clearConfs, useExpTorsionAnglePrefs, useBasicKnowledge, …)
mol:传入mol对象
randomSeed:随机种子,方便结果重复
clearConfs:清除已有构象,默认True
useExpTorsionAnglePrefs和useBasicKnowledge两个参数即控制是否使用ETKDG,默认都为True
>>> AllChem.EmbedMolecule(m3d, randomSeed=10, useExpTorsionAnglePrefs=False, useBasicKnowledge=False)
>>> Draw.MolToImage(m3d, size=(250,250))
- 查看结果
不知道如何生成图片的可以看这篇文章的第四部分。
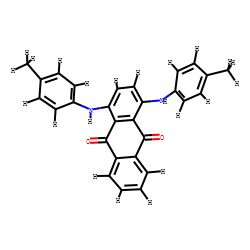
3.距离几何+ETKDG生成3D构象
- 生成3D构象:AllChem.EmbedMolecule()
先用距离几何初始化3D坐标,再使用ETKDG算法优化
参数同上,默认useExpTorsionAnglePrefs和useBasicKnowledge为True
>>> AllChem.EmbedMolecule(m3d, randomSeed=10)
>>> Draw.MolToImage(m3d, size=(250,250))
- 查看结果
比单独使用距离算法确实顺眼了点。
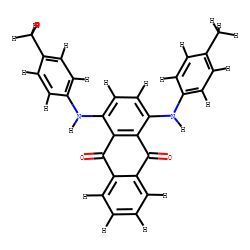
4.距离几何+ETKDG生成多构象
无论使用哪种方法,都可以对一个分子生成许多构象,实现也很简单,无非使用不同的随机种子,多跑几次距离几何的计算过程就行了。
- 生成多个构象:AllChem.EmbedMultipleConfs(mol, numConfs, maxAttempts, randomSeed, clearConfs, pruneRmsThresh, useExpTorsionAnglePrefs, useBasicKnowledge, …)
其中numConfs控制了生成构象的个数。
mol:mol对象
numConfs:生成的构象数量
maxAttempts:尝试生成构象的最多次数
randomSeed:随机种子
clearConfs:清除已有构象
pruneRmsThresh:根据RMS进行合并
ETKDG相关参数同上
>>> cids = AllChem.EmbedMultipleConfs(m3d, numConfs=10)
>>> print(len(cids))
10
- 获取某个构象GetConformer(id)
通过传入id获取指定构象
>>> conf1 = m3d.GetConformer(id=1)
- 计算不同构象间的差异:AlignMolConformers(mol, RMSlist, …)
对同一分子不同的构象先进行重叠排列,再计算RMS(root mean square)值
RMSlist:用于接收RMS的列表,它由第一个构象与剩余9个构象依次比对产生。
>>> rmslist = []
>>> AllChem.AlignMolConformers(m3d, RMSlist=rmslist)
>>> print(len(rmslist))
9
- 也可以计算任意两个指定构象的RMS值GetConformerRMS(mol, confId1, confId2, atomIds, prealigned)
confId1:第一个构象
confId2:第二个构象
atomIds:需要对比的原子,默认全部
prealigned:构象是否已经对齐,默认False(没有时,函数会自动将它们对齐)
>>> AllChem.GetConformerRMS(m2, 1, 9, prealigned=True)
1.5515373147254072
5.距离几何+ETKDG+MMFF生成3D构象
对距离几何产生的构象,进行ETKDG优化后,还可以继续使用MMFF94等力场进行优化。不过需要注意的是,MMFF力场中的原子类型编码采用了自身的芳香性模型,因此在使用MMFF相关方法后,分子的芳香属性(aromaticity flags)会改变。
- 使用MMFF94进行优化构象:MMFFOptimizeMolecule(mol, mmffVariant, maxIters, …)
mmffVariant:可选"MMFF94"或"MMFF94s",默认MMFF94
maxIters:最多迭代次数,默认200
>>> AllChem.EmbedMolecule(m3d, randomSeed=10)
>>> AllChem.MMFFOptimizeMolecule(m3d)
>>> Draw.MolToImage(m3d, size=(250,250))
6.距离几何+ETKDG+MMFF生成多构象
其实当使用ETKDG算法生成3D构象时,通常无需再使用MMFF94力场优化。如果执意要这么做,有一个方便的函数可以调用
- 使用MMFF94优化多个构象:MMFFOptimizeMoleculeConfs()
返回的结果是元组组成的列表,每个元组表示每个构象的收敛值和能量(not_converged, energy)。如果not_converged是0,则收敛,接近最稳态,否则没有到达最稳态。
>>> res = AllChem.MMFFOptimizeMoleculeConfs(m3d)
>>> res
[(0, 102.4284038486388),
(1, 102.42249902079311),...]
7.多线程生成多构象
可以通过numThreads参数进行设置。
- EmbedMultipleConfs(mol, numThreads)
- MMFFOptimizeMoleculeConfs(mol, numThreads)
numThreads:默认为1,表示单进程执行。设置为0表示将会使用本机最大线程数执行。
>>> cids = AllChem.EmbedMultipleConfs(m3d, numThreads=0)
>>> res = AllChem.MMFFOptimizeMoleculeConfs(m3d, numThreads=0)
三、结语
最后需要注意的是,构象生成是一项复杂且漫长的工作。rdkit生成的3D结构并不能取代真实的构象,它仅仅为需要3D结构的场景提供了一种快速实现的方法。不过,rdkit提供的ETKDG方法还是可以满足大多数3D需求场景的。
本文参考自rdkit官方文档。
代码及源文件在这里。