本文主要学习 snakemake--我最喜欢的流程管理工具
- 整合多个流程工具,一个很好的流程框架
- conda 下载
conda install -c bioconda snakemake
snakemake 学习
wget https://bitbucket.org/snakemake/snakemake-tutorial/get/v3.11.0.tar.bz2
tar -xf v3.11.0.tar.bz2 --strip 1
- 看下目录
.
├── README.md
├── data
│ ├── genome.fa
│ ├── genome.fa.amb
│ ├── genome.fa.ann
│ ├── genome.fa.bwt
│ ├── genome.fa.fai
│ ├── genome.fa.pac
│ ├── genome.fa.sa
│ └── samples
│ ├── A.fastq
│ ├── B.fastq
│ └── C.fastq
├── environment.yaml
└── v3.11.0.tar.bz2
1 序列比对
bwa mem data/genome.fa data/samples/A.fastq | samtools view -Sb - > mapped_reads/A.bam 转成snakemake
- Snakefile 文件
# 用你擅长的文本编辑器
vim Snakefile.py
# 编辑如下内容
rule bwa_map:
input:
"data/genome.fa",
"data/samples/A.fastq"
output:
"mapped_reads/A.bam"
shell:
"""
bwa mem {input} | samtools view -Sb - > {output}
"""
-
snakemake -np mapped_reads/B.bam假运行,-n:仅显示执行计划,而不实际执行步骤,-p:打印生成的shell命令以进行说明 - 由于规则有多个输入文件,因此Snakemake会将它们连接起来,并用空格隔开。换句话说,Snakemake将替换
{input}为data/genome.fa data/samples/A.fastq
2 批量比对
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"""
bwa mem {input} | samtools view -Sb - > {output}
"""
- {sample} 代表 A,B,C
- 注意,输出和输入的通配符要相同
- 运行
snakemake -np mapped_reads/{A,B}.bam需要更新之前运行过的文件,让输入文件新与输出文件touch data/samples/A.fastq
3 比对后排序
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample}"
" -O bam {input} > {output}"
- 运行
snakemake sorted_reads/{A,B,C}.bam - T 临时文件
-
wildcards。用来获取通配符匹配到的部分,例如对于通配符"{dataset}/file.{group}.txt"匹配到文件101/file.A.txt,则{wildcards.dataset}就是101,{wildcards.group}就是A。
4. 建立索引和对任务可视化
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
- 运行
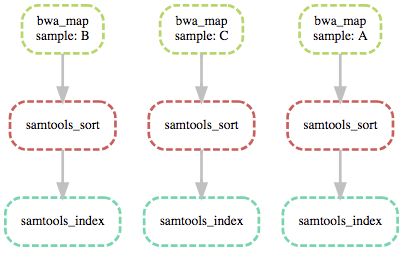
snakemake sorted_reads/{A,B,C}.bam.bai - 流程可视化
snakemake --dag sorted_reads/{A,B,C}.bam.bai | dot -Tsvg > dag.svg
image
5 基因组变异识别
expand("{file}.txt", file=["hello", "world"])
6 编写报告
- 上面都是在规则里执行shell脚本,snakemake的一个优点就是可以在规则里面写Python脚本,只需要把shell改成run,此外还不需要用到引号。
rule report:
input:
"calls/all.vcf"
output:
"report.html"
run:
from snakemake.utils import report
with open(input[0]) as vcf:
n_calls = sum(1 for l in vcf if not l.startswith("#"))
report("""
An example variant calling workflow
===================================
Reads were mapped to the Yeast
reference genome and variants were called jointly with
SAMtools/BCFtools.
This resulted in {n_calls} variants (see Table T1_).
""", output[0], T1=input[0])
- 这里还用到了snakemake的一个函数,report,可以对markdown语法进行渲染生成网页。
基础部分小结
- Snakemake基于规则执行命令,规则一般由
input, output,shell三部分组成。 - Snakemake可以自动确定不同规则的输入输出的依赖关系,根据时间戳来判断文件是否需要重新生成
- Snakemake
以{sample}.fa形式进行文件名通配,用{wildcards.sample}获取sample的实际文件名 - Snakemake用
expand()生成多个文件名,本质是Python的列表推导式 - Snakemake可以在规则外直接写Python代码,在规则内的
run里也可以写Python代码。 - Snakefile的第一个规则通常是
rule all,因为默snakemake默认执行第一条规则
进阶:对流程进一步修饰
- 对于一些工具,比如说bwa,多进程或者多线程运行能够大大加速计算。snakemake使用
threads来定义当前规则所用的进程数,我们可以对之前的bwa_map增加该指令。
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
threads:8
shell:
"bwa mem -t {threads} {input} | samtools view -Sb - > {output}"
2. 配置文件
- 一般存文件路径,软件路径的地方
- 之前的SAMPLES写在了snakefile,也就是意味这对于不同的项目,需要对snakefile进行修改,更好的方式是用一个配置文件。配置文件可以用JSON或YAML语法进行写,然后用configfile: "config.yaml"读取成字典,变量名为config。
- config.yaml 内容为:
samples:
A: data/samples/A.fastq
B: data/samples/B.fastq
- YAML使用缩进表示层级关系,其中缩进必须用空格,但是空格数目不重要,重要的是所今后左侧对齐。上面的YAML被Pytho读取之后,以字典保存,形式为
{'samples': {'A': 'data/samples/A.fastq', 'B': 'data/samples/B.fastq'}}
3. 输入函数
rule bwa_map:
input:
"data/genome.fa",
lambda wildcards: config["samples"][wildcards.sample]
output:
"mapped_reads/{sample}.bam"
threads: 8
shell:
"bwa mem -t {threads} {input} | samtools view -Sb - > {output}"
4. 规则参数
- 有些时候,shell命令不仅仅是由input和output中的文件组成,还需要一些静态的参数设置。如果把这些参数放在input里,则会因为找不到文件而出错,所以需要专门的
params用来设置这些参数。
rule bwa_map:
input:
"data/genome.fa",
lambda wildcards: config["samples"][wildcards.sample]
output:
"mapped_reads/{sample}.bam"
threads: 8
params:
rg="@RG\tID:{sample}\tSM:{sample}"
shell:
"bwa mem -R '{params.rg}' '-t {threads} {input} | samtools view -Sb - > {output}"
5. 日志文件
- 当工作流程特别的大,每一步的输出日志都建议保存下来,而不是输出到屏幕,这样子出错的时候才能找到出错的所在。snakemake非常贴心的定义了log,用于记录日志。好处就在于出错的时候,在log里面定义的文件是不会被snakemake删掉,而output里面的文件则是会被删除。
rule bwa_map:
input:
"data/genome.fa",
lambda wildcards: config["samples"][wildcards.sample]
output:
"mapped_reads/{sample}.bam"
params:
rg="@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
threads: 8
shell:
"(bwa mem -R '{params.rg}' -t {threads} {input} | "
"samtools view -Sb - > {output}) 2> {log}"
高级:实现流程的自动部署
- 我建议你在新建一个snakemake项目时,都先用
conda create -n 项目名 python=版本号创建一个全局环境,用于安装一些常用的软件,例如bwa、samtools、seqkit等。然后用如下命令将环境导出成yaml文件
conda env export -n 项目名 -f environment.yaml
- 全部代码
configfile: "config.yaml"
rule all:
input:
"report.html"
rule bwa_map:
input:
"data/genome.fa",
lambda wildcards: config["samples"][wildcards.sample]
output:
temp("mapped_reads/{sample}.bam")
params:
rg="@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
threads: 8
shell:
"(bwa mem -R '{params.rg}' -t {threads} {input} | "
"samtools view -Sb - > {output}) 2> {log}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
protected("sorted_reads/{sample}.bam")
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=config["samples"]),
bai=expand("sorted_reads/{sample}.bam.bai", sample=config["samples"])
output:
"calls/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule report:
input:
"calls/all.vcf"
output:
"report.html"
run:
from snakemake.utils import report
with open(input[0]) as vcf:
n_calls = sum(1 for l in vcf if not l.startswith("#"))
report("""
An example variant calling workflow
===================================
Reads were mapped to the Yeast
reference genome and variants were called jointly with
SAMtools/BCFtools.
This resulted in {n_calls} variants (see Table T1_).
""", output[0], T1=input[0])