STAMP:扩增子、宏基因组统计分析神器(中文帮助文档)
- 简介
- 软件简介
- STAMP能干什么
- 分析实战
- 输入文件
- 多组比较——肠型
- 属性面板功能简介
- 图表类型介绍
- 统计表导出
- 其它功能
- 统计方法
- 关于样本重复
- 多组比较
- 分析两组
- 分析两样品
- 常见问题
- 读入文件错误
- 实验设计和丰度矩阵样品名不对应
- 总结
- Reference
- 猜你喜欢
- 写在后面
之前本平台分享过STAMP的使用:
- 微生物组间差异分析神器-STAMP
今天带来了更详细、深入的讲解和高级玩法。
简介
软件简介
STAMP是一款用于分析微生物分类和功能谱的软件,不仅可以做统计,更能绘制多种图形,方便发表使用。
1.0于2010年发表于Bioinformatics,被引493次;2.0于2014年1月发表于Bioinformatics,引用483次(截止2018年5月26日)。
官网地址:http://kiwi.cs.dal.ca/Software/STAMP
最新版本2.1.3,Downloads部分可以下载适合自己系统:如Windows/Linux/MacOS版本的软件。本文内容主要翻译自STAMP用户手册v2,2014年12月15日版本 http://kiwi.cs.dal.ca/Software/images/c/cd/STAMP_Users_Guide.zip ,读者添加了自己的理解,以及按读者自己的学习逻辑重排。一些章节标注了原文页码,如P4代表原文第4页。
安装目录中Examples目录提供了示示例分析结果,以及演示数据实例。
STAMP能干什么
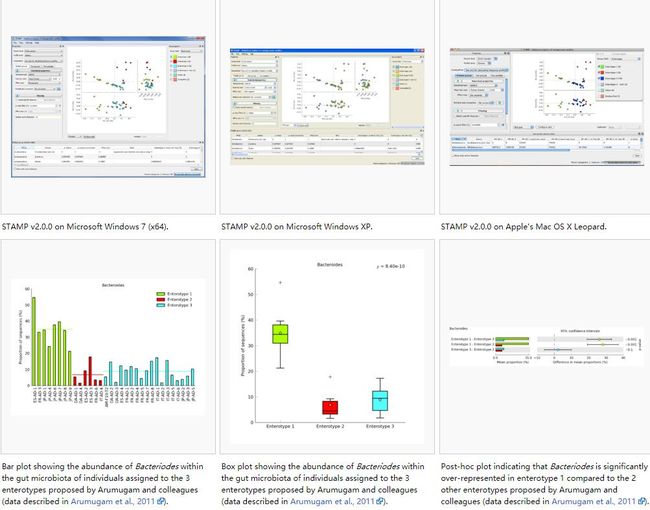
STAMP可以现实不同平台下兼容性分析数据,主要包括Beta多样性散点图、物种丰度柱状图、箱线图,以及Post-hoc图展示差异物种/功能。
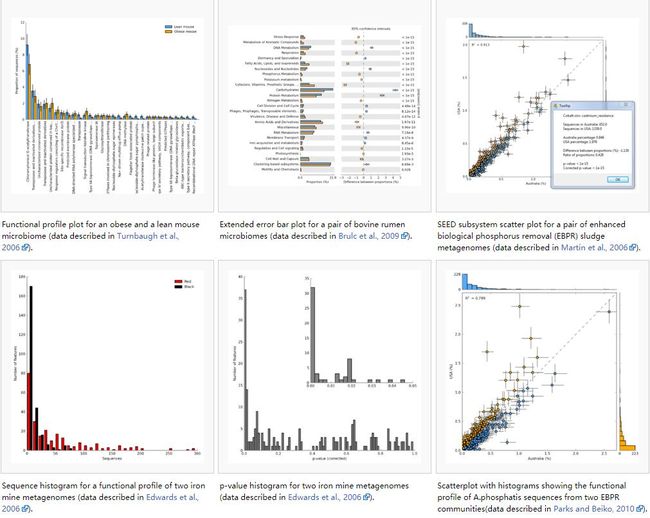
还可以绘制带误差线柱状图、误差线和柱分离组合图、相关散点图、密度柱状图、P值柱状图等统计图表。
分析实战
输入文件
本质上是文本的OTU表或功能组成矩阵 + 实验设计即可。同时也支持biom格式。MG—RAST、IMG/M、CoMet、RITA等软件的结果。
OTU表/功能组成矩阵
最常见的是OTU表、各分类级物种组成;宏基因组的物种和功能组成表/矩阵。支持多列特征分级属性,但必须是严格的等级。多级时容易报错,通常只使用1或2级行名,如下表门和属对应样品中相对丰度。
Phyla Genera AM-AD-1 AM-AD-2 AM-F10-T1 AM-F10-T2
Bacteroidetes Bacteroides 9.7172748 5.248866 1
Chlorobi Prosthecochloris 0.0 0.0 0.0 0.0 0.0 0
Chloroflexi Chloroflexus 0.0 0.0 0.0 0.0 0.0 0.0 0
Chloroflexi Dehalococcoides 0.0 19.9791942 2.3873250
实验设计Metadata
一定要包括样品名和组名,可以有多种分组方式
Sample Id Enterotype Nationality Clinical Status Gender Project Clinical Status [filtered] Nationality [filtered] Gender [filtered]
AM-AD-1 Unclassified american healthy F gill06 na na na
AM-AD-2 Unclassified american healthy M gill06 na na na
多组比较——肠型
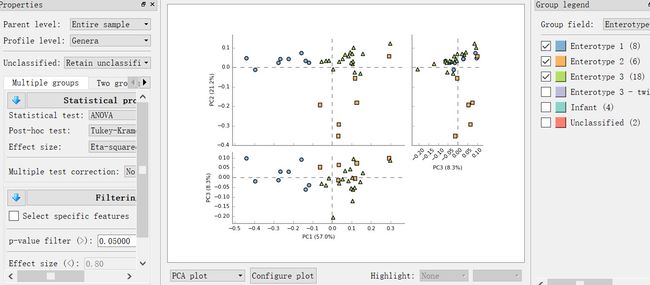
安装好软件,Load data打开example中的EnterotypesArumugam目录中的肠型数据(spf是数据矩阵,tsv是实验设计),打开默认显示PCA结果如下:
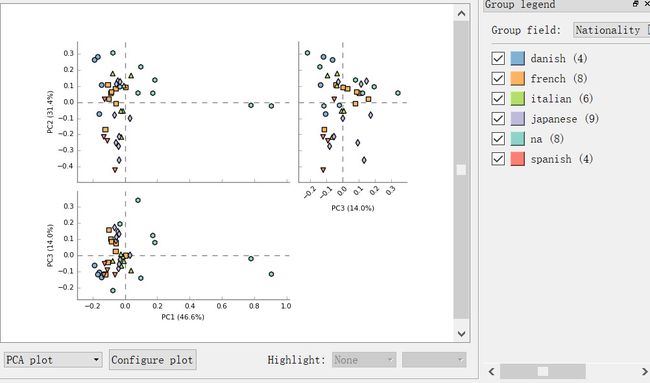
展示PC1-3之间组合的散点图。图片大小、关闭其它轴、图例位置等可以下方Configure plot中设置,图例在右侧,每组不同颜色,可在Group field中选择实验设计中不同的分组,分组可通过勾选进行取消或选择,并实时显示分析结果。(大数据时,请点击右下角实时计算,减少等待时间)
重现肠型分类
选择右上角Group field为Enterotype,去除后三个非主要分组,只保留三种肠型;同时左侧的数据属性中,Profile level选择Genera,现在我们可以看到三种肠型的不同形状在图中分开比较明显。
属性面板功能简介
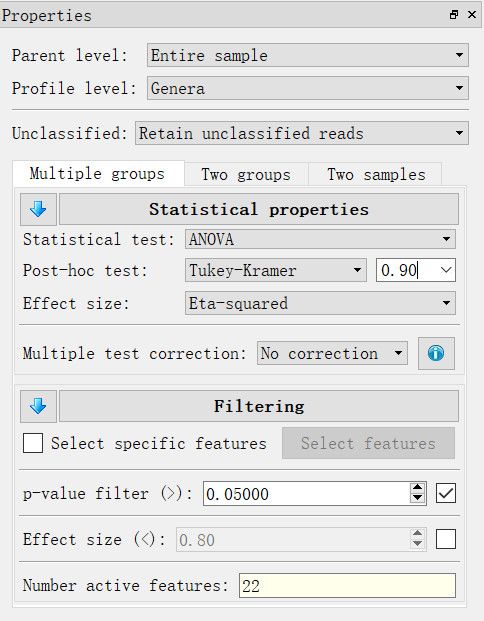
- Parent level:标准化的总体范围
- Profile level: 分析的具体级别,如科、属、种或OTU
- Unclassified: 末分类数据处理方法,分别为Retain保留、Remove移除和仅用于计算比例。不同处理方法,结果会很大差异。
- Statistical properties:统计属性,主要包括统计检验方法,事后检验方法和置信区间,效应大小,多重检验校正方法等的选择;
- Filtering: 过滤阈值,主要是设置P值和效应大小。修改后,下方同步有符合条件的结果数量。方便在查看图表结果时只关注符合条件的features。
图表类型介绍
比较常用的是两组比较,本软件对多组比较支持也非常好,很容易进一步探索数据。
- 柱状图:显示每个样品中feature的相对比例,并添加组均值,方便查看单个Feature的数据分布,如下图显示三种肠型中拟杆菌属的相对丰度。
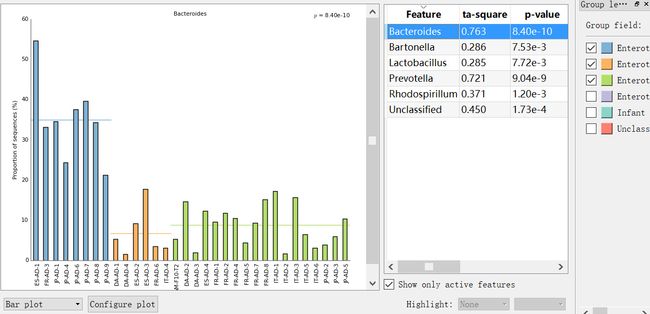
注:Feature列表下方,可勾远Show active来只显示符合条件的结果。
- 箱线图:简单快速显示组内数据分布。
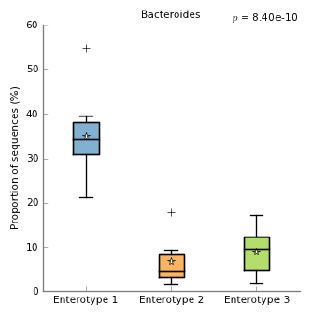
箱线图展示三种肠型中拟杆菌的相对丰度分布和整体统计P值
具体解析,可阅读 - 扩增子图表解读-理解文章思路
- 热图:显示每个Features在样品中丰度的比例,优热在于不仅显示所有样本的丰度值,更可以对行Features和列样品进行聚类显示之间的关系;
- 主成分分析PCA:散点图在低维空间显示高维数据间主要差异;
- Post-hoc图:多组统计检验的无效假设(如ANOVA或Kruskal-Wallis)是所有组相等。提供每对组间测量的P-value和效应大小。

扩展柱状图/事后图显示组间两两比较柱状图,及置信区间分布和P值。
图片的具体参数见Configure plot页面,可在File菜单中Save plot保存图片,有PNG位图,和PDF, PS, EPS, SVG共4种矢量图可选,推荐PDF格式方便查看和修改。
统计表导出
我们想导出统计结果的表格,方便发表文章作为原始数据,和进一步分析。
View —— Multiple group statistics table
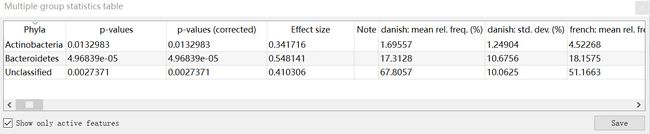
可以看到特征Feature、P值、假阳性率FDR、效应大小、各组均值和标准差等。可选择全部或显示部分,Save保存表格至文件。(P12)
其它功能
全局设置:Setting - Preferences中可设置丰度过滤、bootstrap次数、图标签截短长度、轴颜色、其它样本颜色、和P-value阈值等
软件的扩展:可编写添加新统计方法和图表类型
统计方法
关于样本重复
需要多少样本才能检测统计显著?推荐阅读:Suresh and Chandrashekara (2012) and the article “Getting the Sample Size Right” by Jeremy Miles (http://www.jeremymiles.co.uk/misc/power/)。
我们的建议是没有最小的样本数,但统计假设必须符合数据分布。小样本量更可能不符合假设。小样本量也不太可能有足够的统计强度来鉴定效应大小。如t-test考虑每组4个样本,可以提供足够的准确度和精确度的信息。
样本数量也由样品本身决定,如果原始样本稳定且测量结果精度高,只需极少样本可决定统计差异,如不同面值硬币间重量差异,因为它们制作的精度高且准确称量较容易。
生物学数据充满了各种噪音,物种和功能组成也容易受多种因素影响,具有低准确度和精密度的特点。实验和分析每步都可能对想结果有影响,因此我们需要生物学重复,才能鉴定组间差异。如健康和疾病样本,有多种因素不可控,需要极大量样本才可能发现均值和方差显著的不同。(P6、原文Page|6)
多组比较
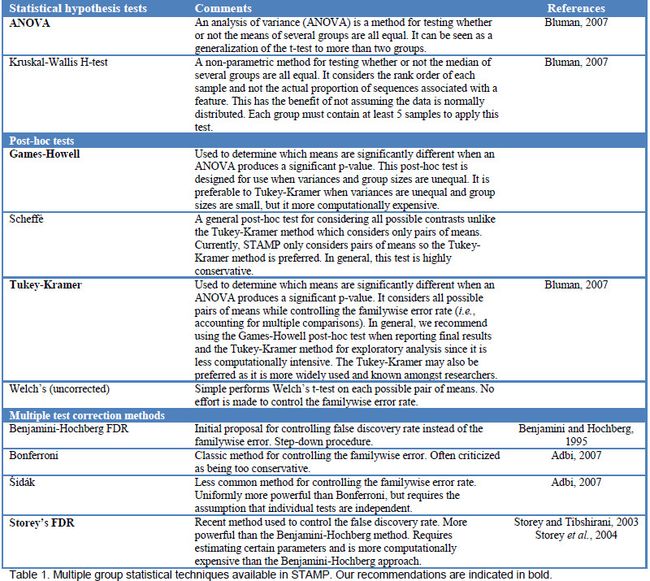
| 统计假设方法 | 描述 |
|---|---|
| ANOVA | 方差分析(analysis of variance)的缩写,用于检验多组均值是否相等的方法。可被认为是可分析多组的t-test |
| Kruskal-Wallis H-test | 无参数的秩合检验方法,检验多组的中位数是否相等。它考虑样品排序位置而不是真实数值或比例。它不基于数据是正态分布的前提。此方法要求每组至少5个样本。 |
| 事后检验 | 描述 |
|---|---|
| Games-Howell | 当ANOVA产生了显著P值后,检验具体哪两个均值显著不同。用于组样本和方差不同。当方差不同,组样本量小时推荐使用Tukey-Kramer方法 |
| Scheffe | 考虑所有可能的比较,而Tukey-Kramer只考虑成对均值。此种方法较保守 |
| Tukey-Kramer | 用于ANOVA显著后进一步成对比较。考虑所有可能的均值队,并考虑多次比较的错误率控制。推荐使用Games-Howell输出最终结果,而Tukey-Kramer用于探索分析。推荐此方法的另一个原因是此法使用广泛,被研究者所熟知。 |
| Welch’s(uncorrected) | 只是成队均值比较,但不进行多次比较的错误率控制 |
| 多重检验校正方法 | 描述 |
|---|---|
| Benjamini-Hochberg FDR | 控制假阳性率FDR |
| Bonferroni | 控制整体错误率的经典方法,被批评太保守 |
| Sidak | 在整体错误率控制中使用不多,但均匀分布数据上比Bonferroni更强,但需要假设个体检验是独立的 |
| Storey’s FDR | 控制FDR的新方法,比BH更强。需要估计一些参数和更多的计算资源。 |
表1. STAMP中可用的多组比较方法,其中加粗为推荐方法(STAMP官方帮助文档P14)
分析两组


| 统计假设方法 | 描述 |
|---|---|
| t检验 | T检验,亦称student t检验(Student’s t test),假设两组有相同的方差,当假设成立时,它比Welch’s检验更强,主要用于样本含量较小(例如n<30),总体标准差σ未知的正态分布。 |
| Welch’s t-test | t-test的一种变形,用于当两组无法满足方差相同的假设时使用。 |
| White’s无参t-test | 无参数的检验,由White为临床宏基因组数据分析提出。此方法使用排序过程移除标准t-test的正态假设。此外,它使用启法式鉴定松散的特征,可采用Fisher精确检验和pooling的策略,适合组样本一致,或小于8个样品。大数据集计算耗时。 |
| 置信区间方法 | 描述 |
|---|---|
| DP: t-test inverted | 只有当方差相等的t检验可用。 |
| Scheffe | 考虑所有可能的比较,而Tukey-Kramer只考虑成对均值。此种方法较保守 |
| DP: Welch’s inverted | 为Welch’s t检验提供置信区间。 |
| DP: bootstrap | 适合White’s 无参t-test |
| 多种检验校正方法 | 描述 |
|---|---|
| Benjamini-Hochberg FDR | 控制假阳性率FDR |
| Bonferroni | 控制整体错误率的经典方法,被批评太保守 |
| Sidak | 在整体错误率控制中使用不多,但均匀分布数据上比Bonferroni更强,但需要假设个体检验是独立的 |
| Storey’s FDR | 控制FDR的新方法,比BH更强。需要估计一些参数和更多的计算资源。 |
表2. STAMP中可用的多组比较方法,其中加粗为推荐方法(STAMP官方帮助文档P17)
分析两样品
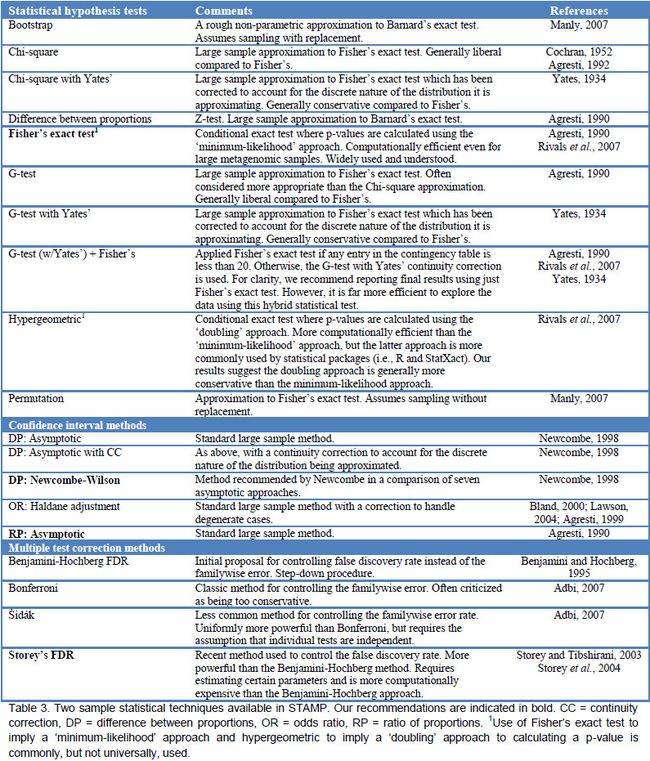
| 统计假设方法 | 描述 |
|---|---|
| Bootstrap | 一种无参方法,与Barnard精确检验相似,假设放回抽样 |
| 卡方Chi-squre | 大样本与Fisher精确检验类似,但更自由 |
| Yates卡方 | 在卡方基础上考虑了分布,比Fisher更保守 |
| Fisher精确检验 | 条件精确检验,P值采用最大似然方法。宏基因组大数据样本计算速度快,应用广泛且公众认可 |
| G-test | 大样本与Fisher近似,比卡方更合适,比Fisher更灵活 |
| G-test with Yates’ | 大样本与Fisher类似,考虑自然离散校正,比Fisher更保守 |
| G-test(w/Yates’)+Fisher’s | 当列联表中小于20使用Fisher精确检验,其它使用G-test。为了结果清楚,我们推荐只使用Fisher精确检验。而在探索数据阶段,使用混合的统计方法可能更有效 |
| 超几何分布 | P值使用两种方法的条件精确检验。比最小似然法(在R和StatXact中常用)更快。但更保守。 |
| 置换 | 与Fisher类似,假定无放回抽样 |
| 置信区间方法 | 描述 |
|---|---|
| DP: 渐近 | 标准的大样本方法 |
| Scheffe | 考虑所有可能的比较,而Tukey-Kramer只考虑成对均值。此种方法较保守 |
| DP: CC渐近 | 考虑自然离散分布和连续校正 |
| DP: Newcombe-Wilson | Newcombe推荐的7种渐近方法中最优的 |
| OR: Haldane adjustmet | 大样本方法结合校正解决退化问题 |
| RP: 渐近 | 标准的大样本方法 |
| 多重检验校正方法 | 描述 |
|---|---|
| Benjamini-Hochberg FDR | 控制假阳性率FDR |
| Bonferroni | 控制整体错误率的经典方法,被批评太保守 |
| Sidak | 在整体错误率控制中使用不多,但均匀分布数据上比Bonferroni更强,但需要假设个体检验是独立的 |
| Storey’s FDR | 控制FDR的新方法,比BH更强。需要估计一些参数和更多的计算资源。 |
表3. STAMP中两样品统计方法。推荐方法加粗。CC = 连续校正,DP = 比例差异,OR = 让步比,RP = 比例。
常见问题
读入文件错误
Data does not form a strick hierarchy. Child Unassigned has multiple parents.
它要求的输入分类级不允许在各级别有重名,而物种命名总有些不规范的级别和末命名的,所以建议将门、纲、目、科、属、OTU水平分别制作成spf文件给STAMP分析,可确保正常使用;
STAMP也提供了检查不符合要求的,非层级的分类检查脚本
wget http://kiwi.cs.dal.ca/Software/images/e/e6/CheckHierarchy.zip
unzip CheckHierarchy.zip
chmod +x checkHierarchy.py
# 一个使用RDP注释结果,发现4419条非层级结果,我想哭,完全无法使用
checkHierarchy.py result/otutab_stamp.spf | wc -l
# 检查结果如下
checkHierarchy v0.0.1:
by Donovan Parks ([email protected])
Identified 3880 samples.
Identified 8 hierarchical columns.
The following entries have two (and potentially more) parents:
12 Order Actinomycetales Actinobacteria,Alphaproteobacteria
35 Family Rhodospirillaceae Actinomycetales,Rhodospirillales
38 Family Sphingobacteriaceae Sphingobacteriales,Cytophagales
54 Order Clostridiales Clostridia,Acidobacteria_Gp21
末注释的最好统计标记为unclassified,上文提到有多种处理方法,但没有完美解决问题。
实验设计和丰度矩阵样品名不对应
Metadat warnings: Missing metadat for the following samples:
实验设计中缺失OTU表中的样品名,如果是人为注释或去除掉的,可以忽略此警告,否则仔细检查实验设计是否与矩阵中样品名对应
总结
分析三步曲:
整体:PCA plot,可通过不断筛选分组来观察组间整体差异
多组: 组间重复数3-15个,可用bar或boxplot逐个查看显著差异的OTUs,大于15个最好只用boxplot;组内样本波动大用boxplot更直观,波动小可选barplot+error bar也很漂亮。组间差异明显,组内重复好,可选热图+聚类信息更丰富。
两组:boxplot,barplot用原始数据。整体可用extended barplot
软件功能非常强大,但还是些局限性:如有些步骤不能选择原始数据统计;强行进行了标准化,这样对部分数据分析可能会有影响;软件无法保存工作状态、上次访问路径等;新项目必须关闭软件重新打开才能开始分析等。
Reference
- Parks DH and Beiko RG. (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics, 26, 715-721.
- Parks DH, Tyson GW, Hugenholtz P, Beiko RG. (2014). STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics, 30, 3123-3124.
- 微生物组间差异分析神器-STAMP
- STAMP:“花样”分析组间差异
猜你喜欢
- 10000+:肠道细菌 人体上的生命 宝宝与猫狗 梅毒狂想曲 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊
- 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板 Shell R Perl
- 生物科普 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外1500+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”

点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA