QIIME 2用户文档. 6沙漠土壤分析Atacama soil(2018.11)
文章目录
- 前情提要
- QIIME 2用户文档. 6阿塔卡马沙漠微生物组分析
- 启动QIIME2运行环境
- 实验数据下载
- 双端数据分析方法
- 去噪并生成特征表和代表序列
- 接下来分析要回答的科学问题
- Reference
- 译者简介
- 猜你喜欢
- 写在后面
前情提要
- 文章导读:QIIME 2可重复、交互和扩展的微生物组数据分析流程
- 1简介和安装
- 2插件工作流程概述
- 3老司机上路指南
- 4人体各部位微生物组分析
- 5粪菌移植分析练习
QIIME 2用户文档. 6阿塔卡马沙漠微生物组分析

原文地址: https://docs.qiime2.org/2018.11/tutorials/atacama-soils/
此实例需要一些基础知识,要求完成本系列文章前两篇内容:《1简介和安装》和《4人体各部位微生物组分析》。
本教程设计用于两个目的。首先,它描述了对双端序列分析的初始处理步骤,直到分析步骤与单端序列分析相同。这包括导入、样本拆分和去噪步骤,并产生特征表和相关的特征序列。其次,这是一次自我练习,可以在《4人体各部位微生物组分析》之后运行,以获得更多使用QIIME 2的经验。对于这个练习,我们提供了一些可以用来指导分析的问题,但是不提供直接解决每个问题的命令。相反,您应该应用您在《4人体各部位微生物组分析》中学到的命令。
在本教程中,您将使用QIIME 2对来自智利北部阿塔卡马沙漠的土壤样本进行分析。阿塔卡马沙漠是地球上最干旱的地方之一,有些地区每十年降雨量不到一毫米。尽管极端干旱,土壤中仍然有微生物。本研究采样地点为东部的巴克达诺(Baquedano)和西部的永盖(Yungay),横断面的平均土壤相对湿度与海拔高度呈正相关(海拔越高,干旱程度越轻,平均土壤相对湿度越高)。沿着这些剖面,在每个地点挖坑,从每个坑的三个深度收集土壤样品。
启动QIIME2运行环境
对于上文提到了两种常用安装方法,我们每次在分析数据前,需要打开工作环境,根据情况选择对应的打开方式。
# 创建qiime2学习目录并进入
mkdir -p qiime2
cd qiime2
# Miniconda安装的请运行如下命令加载工作环境
source activate qiime2-2018.11
# 如果是docker安装的请运行如下命令,默认加载当前目录至/data目录
# docker run --rm -v $(pwd):/data --name=qiime -it qiime2/core:2018.11
# 创建本节学习目录
mkdir qiime2-atacama-tutorial
cd qiime2-atacama-tutorial
实验数据下载
Obtain the data
注意:QIIME 2 官方测试数据部分保存在Google服务器上,国内下载比较困难。可使用代理服务器(如蓝灯)下载,或公众号后台回复"qiime2"获取测试数据批量下载链接,你还可以跳过以后的wget步骤。
下载来源Google文档的实验设计
wget \
-O "sample-metadata.tsv" \
"https://data.qiime2.org/2018.11/tutorials/atacama-soils/sample_metadata.tsv"
下载双端实验数据(使用10%抽样数据方便下载和演示):分别为正向、反向和barcodes序列三个文件;文来自亚马逊云,有时无法下载或断开,可下载同一个文件不同时间多试几次就成功了。
mkdir emp-paired-end-sequences
wget \
-O "emp-paired-end-sequences/forward.fastq.gz" \
"https://data.qiime2.org/2018.11/tutorials/atacama-soils/10p/forward.fastq.gz"
wget \
-O "emp-paired-end-sequences/reverse.fastq.gz" \
"https://data.qiime2.org/2018.11/tutorials/atacama-soils/10p/reverse.fastq.gz"
wget \
-O "emp-paired-end-sequences/barcodes.fastq.gz" \
"https://data.qiime2.org/2018.11/tutorials/atacama-soils/10p/barcodes.fastq.gz"
双端数据分析方法
Paired-end read analysis commands
双端数据导入,数据建库类型为EMP双端序列EMPPairedEndSequences(本示例来自EMP项目)
qiime tools import \
--type EMPPairedEndSequences \
--input-path emp-paired-end-sequences \
--output-path emp-paired-end-sequences.qza
输出对象:
- emp-paired-end-sequences.qza: EMP项目双端测序类型
按Barcode序列信息进行样品拆分:--m-barcodes-file为含有样品与barcode信息对应的实验设计,--m-barcodes-category指定含有barcode信息的列名称,--i-seqs输入文件,--o-per-sample-sequences输出文件, --p-rev-comp-mapping-barcodes为barcode方向类型,可用实验设计的barcode与测序文件中的barcode比对以确定方向。本次分析中为反向互补类型。拆分后对拆分样品的结果和质量进行统计。
qiime demux emp-paired \
--m-barcodes-file sample-metadata.tsv \
--m-barcodes-column BarcodeSequence \
--i-seqs emp-paired-end-sequences.qza \
--o-per-sample-sequences demux.qza \
--p-rev-comp-mapping-barcodes
qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
输出对象:
- demux.qza: 样品拆分结果文件
输出可视化:
- demux.qzv: 样本拆分结果可视化
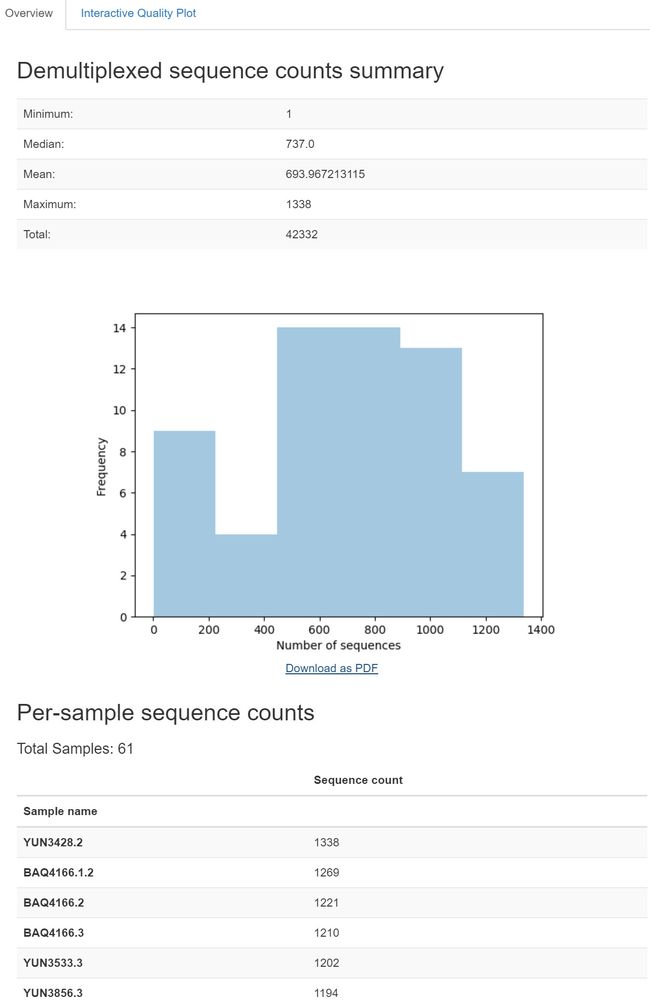
图1. 数据量汇总图表。中位数有737,可以分析练手了。
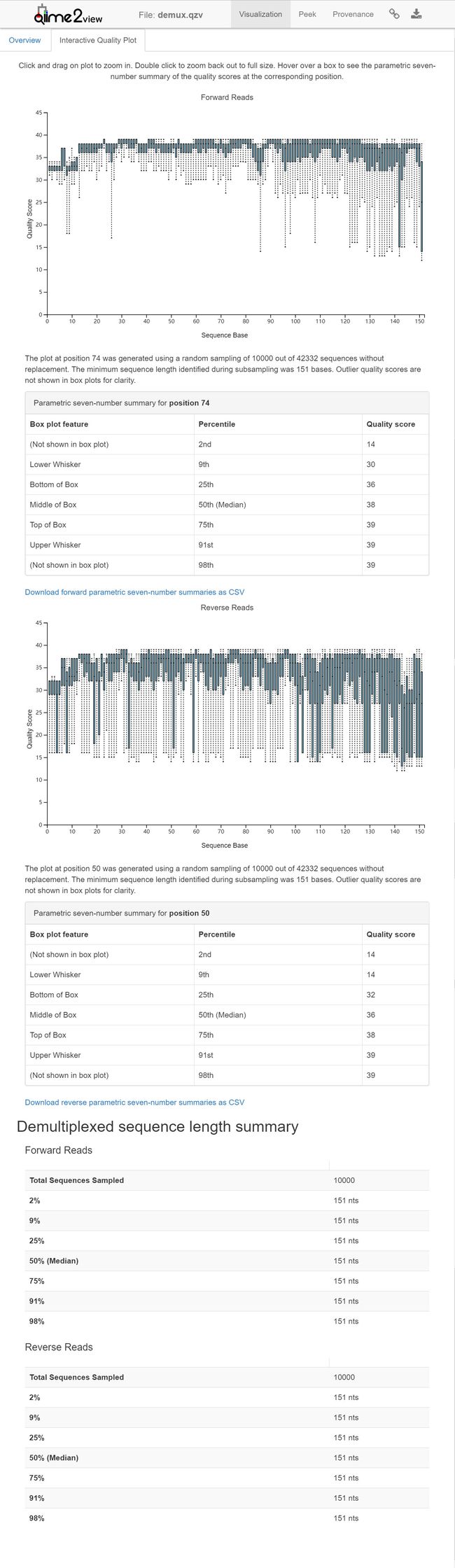
图2. 双端数据质量评估图。
网页中交互式图形可以查看每个碱基位置的详细信息。质量分析后,我们根据上图结果和相关表格来确定下步denoise分析参数。
去噪并生成特征表和代表序列
在序样本拆分之后,我们将基于十个随机选择的样本来查看序列质量,然后对数据进行去噪。当您查看质量图表时,请注意,与《4人体各部位微生物组分析》中的对应图表相比,现在每个示例有两个图表。左边的图表显示正向读取的质量分数,右边的图表显示反向读取的质量分数。我们将使用这些图来确定要使用DADA2进行去噪的裁剪参数,然后使用dada2对双端序列进行去噪。
在这个例子中,我们有150个碱基的正向和反向序列。因为我们需要序列足够长的重叠,以便双端序列可以连接,所以正向和反向序列的前13个基数被修剪,但是没有对序列的末端进行修剪,以避免将读数长度减少太多而无法重叠连接。在这个示例中,对--p-trim-left-f和--p-trim-left-r以及--p-trunc-len-f和--p-trunc-len-r提供了相同的值,但这不是必需的。
qiime dada2 denoise-paired \
--i-demultiplexed-seqs demux.qza \
--p-trim-left-f 13 \
--p-trim-left-r 13 \
--p-trunc-len-f 150 \
--p-trunc-len-r 150 \
--o-table table.qza \
--o-representative-sequences rep-seqs.qza \
--o-denoising-stats denoising-stats.qza
输出对象:
- denoising-stats.qza: 去噪过程统计
- rep-seqs.qza: 代表序列
- table.qza: 特征表
我们要对获得的表和序列进行统计
# 查看Feature/OTU表的统计结果
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv
# 代表序列统计
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
可视化结果:
- table.qzv: 特征表统计
- rep-seqs.qzv: 代表序列统计
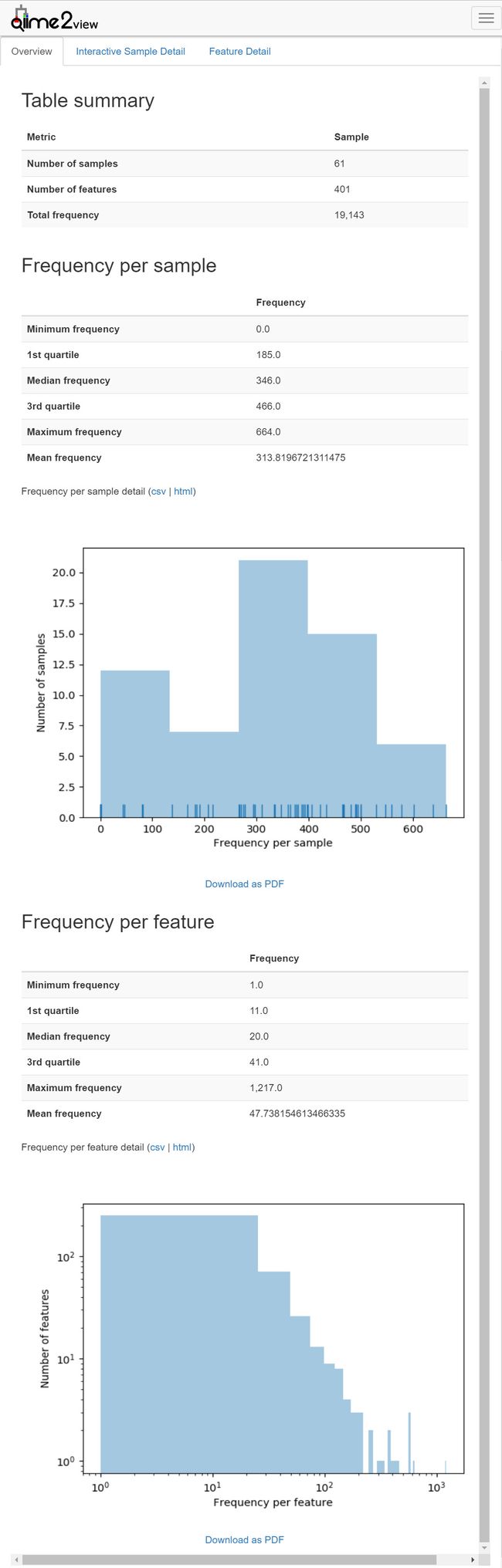
图3. 特征表统计统计,我们要根据数据量,来选择合适的重采样值

图4. 代表性序列统计,长度基本全一致,意义不大。可以点击序列查询相关注意比较方便。
也可以可视化去噪结果:
qiime metadata tabulate \
--m-input-file denoising-stats.qza \
--o-visualization denoising-stats.qzv
输出可视化结果:
- denoising-stats.qzv: 去噪过程统计可视化
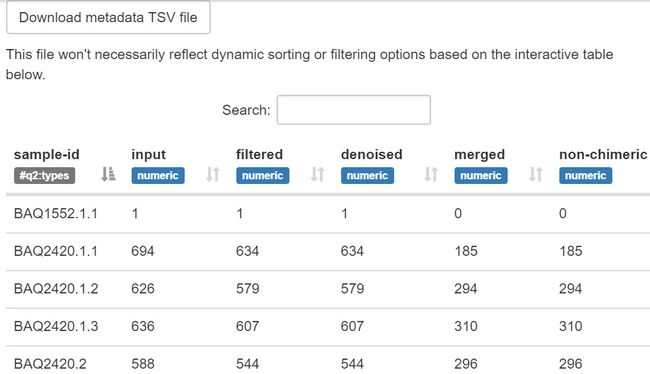
图5. 去噪过程统计,可以看各阶段数据剩余的量。双端合并阶段数据是极速下降的。
接下来,分析双端序列和之前的单端序列就一样了。我们可以继续按照《4人体各部位微生物组分析》中的命令继续分析啦!
接下来分析要回答的科学问题
Questions to guide data analysis
通过以下问题,来指导你分析数据。
- 接下来特征表重采样标准化参数
--p-sampling-depth应该选多少?基于你重采样的参数,有多少样品应该从实验中剔除?在core-metrics-phylogenetic分析中,使用过滤后的样本有多少数据量? - 实验设计中的那种分组方式下微生物组成差异最大?采用那种距离计算方法分开更明显,是
unweighted UniFrac还是Bray-Curtis?根据你对这些距离计算方法的理解,这些不同代表什么意义呢?对于连续型的样本属性,考虑尝试使用qiime metadata distance-matrix与qiime diversity mantel和qiime diversity bioenv结合使用更有效,这些命令之前没有提到过,但可以使用--help查看详细帮助。 - 分析样本连续型属性与样本的丰富多、均匀度之间的关系?推荐使用
qiime diversity alpha-correlation分析多样性与样本属性间的相关性,看看能得到什么结论?不会记得查看帮助文档。 - 哪种样本的分类与Alpha多样性差异最相关,并比较是否有显著差异?
- 在门水平查看不同土壤相对温度下微生物组成,哪个门丰度最高?看那些种类与湿度正/负相关?
- 在有无植被的取样地点,什么菌门差异明显?
Reference
- https://docs.qiime2.org/2018.11/tutorials/atacama-soils/
- mSystems. 2017 May 30;2(3). pii: e00195-16. doi: 10.1128/mSystems.00195-16. eCollection 2017 May-Jun. Significant Impacts of Increasing Aridity on the Arid Soil Microbiome. Neilson JW1, Califf K2, Cardona C3, Copeland A1, van Treuren W4, Josephson KL1, Knight R5, Gilbert JA6, Quade J7, Caporaso JG2, Maier RM1. DOI: 10.1128/mSystems.00195-16 https://www.ncbi.nlm.nih.gov/pubmed/28593197
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope E, Da Silva R, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley G, Janssen S, Jarmusch AK, Jiang L, Kaehler B, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MG, Lee J, Ley R, Liu Y, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton J, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson, II MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CH, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG. 2018. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints 6:e27295v2 https://doi.org/10.7287/peerj.preprints.27295v2
译者简介
刘永鑫,博士。2008年毕业于东北农大微生物学专业。2014年中科院遗传发育所获生物信息学博士学位,2016年博士后出站留所工作,任宏基因组学实验室工程师,目前主要研究方向为宏基因组学、数据分析与可重复计算和植物微生物组、QIIME 2项目参与人。发于论文12篇,SCI收录9篇。2017年7月创办“宏基因组”公众号,目前分享宏基因组、扩增子原创文章300+篇,代表博文有《扩增子图表解读、分析流程和统计绘图三部曲》,关注人数3万+,累计阅读400万+。
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2600+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA