Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients
文章: 新冠肺炎患者支气管肺泡灌洗液与外周血单核细胞的转录组学特征
期刊: SSRN
影响因子: NA
材料: COVID-19患者的支气管肺泡灌洗液和外周血单核细胞分离的RNA
技术: RNA-seq
分享人: 赖为洁
摘要
COVID-19持续爆发,对公共卫生构成了巨大威胁。但是,我们对这种致命疾病的宿主免疫反应的了解有限,是我们无法判断有效的支持治疗药物和治疗方法。
为了获取宿主对SARS-CoV-2(HCoV-19)感染的炎症反应的转录特征,我们对从COVID-19患者的支气管肺泡灌洗液(BALF)和外周血单核细胞(PBMC)标本中分离的RNA进行了转录组测序。
研究结果揭示了患者SARS-CoV-2感染的不同宿主炎症细胞因子谱,并揭示了COVID-19发病与细胞因子过度释放(例如CCL2 / MCP-1,CXCL10 / IP-10,CCL3 / MIP-1A以及CCL4 / MIP1B)之间的关联。
此外,SARS-CoV-2诱导淋巴细胞凋亡和P53信号通路的激活可能是患者淋巴细胞减少的原因。
COVID-19患者的转录组数据集将为抗炎药物的临床指导和了解宿主反应的分子机制提供宝贵的资源。
材料
3例SARS-CoV-2患者和3例健康供体获得了全血和PBMC(Ficoll制剂)
方法
1.RNA-seq reads were firstly mapped to rRNA sequences to remove potential rRNA reads using STAR (v2.7.2b)with the default parameter.
首先使用默认参数STAR(v2.7.2b)将RNA-seq reads 映射到rRNA序列,以去除潜在的rRNA reads。(寡聚-dT(oligo-dTs )共价偶联的磁珠纯化信使RNA)
2.The rest reads were then mapped to the human genome (hg38) with GENCODE gene annotation (v32) with parameters “–sjdbScore 1 –outFilterMultimapNmax 20 –outFilterMismatchNmax 999 –outFilterMismatchNoverReadLmax 0.04 –alignIntronMin 20 –alignIntronMax 1000000 –alignMatesGapMax 1000000 –alignSJoverhangMin 8 –alignSJDBoverhangMin 1” following the guideline of ENCODE RNA-seq pipeline (https://github.com/ENCODE-DCC/long-rna-seq-pipeline).
将其余的reads通过GENCODE基因注释(v32)映射到人类基因组(hg38),参数为“ –sjdbScore 1 –outFilterMultimapNmax 20 –outFilterMismatchNmax 999 –outFilterMismatchNoverReadLmax 0.04 –alignIntronMin 20 –alignIntronMax 1000000 –alignMatesGapMax 1000000 –alignSJoverhangMin 8 ”遵循ENCODE RNA-seq管道指南(https://github.com/ENCODE-DCC/long-rna-seq-pipeline)。
- The human un-mappable reads were then mapped to the SARS-CoV-2 genome as previously reported. PCR replicates mapped in the human genome were removed with picard MarkDuplicates program (v2.13.2-1) . RNA-seq signal tracks were generated by using bam2wig.py provided in RSeQC package (v3.0.0), and visualized in UCSC genome browser with custom track hub.
然后,将不能比对到人的reads定位到SARS-CoV-2基因组。用picard MarkDuplicates程序(v2.13.2-1)删除了映射在人类基因组中的PCR复制。
通过使用RSeQC软件包(v3.0.0)中提供的bam2wig.py生成RNA-seq信号轨道,并在UCSC基因组浏览器中使用自定义轨道中心对其进行可视化。
4.Gene expression was calculated by featureCounts in SubReads package (v1.5.3) with the “-M” parameter. Differentially expressed genes were called by using DESeq2 package (v1.26.0) with the following criteria: adjusted p-value < 0.05 and fold-change > 2. The expressed genes (requiring reads counts greater than 10 and 100 for BALF and PBMC, respectively), were selected and normalized to counts per millions (CPM) for further analysis.
基因表达是通过SubReads软件包(v1.5.3)中的featureCounts 使用“ -M”参数来计算的。
通过使用DESeq2程序包(v1.26.0)调用差异表达的基因,其标准如下:调整后的p值<0.05,倍数变化>2。
选择表达的基因(分别要求BALF和PBMC的读数分别大于10和100),并将其标准化为每百万计数(CPM)进行进一步分析。
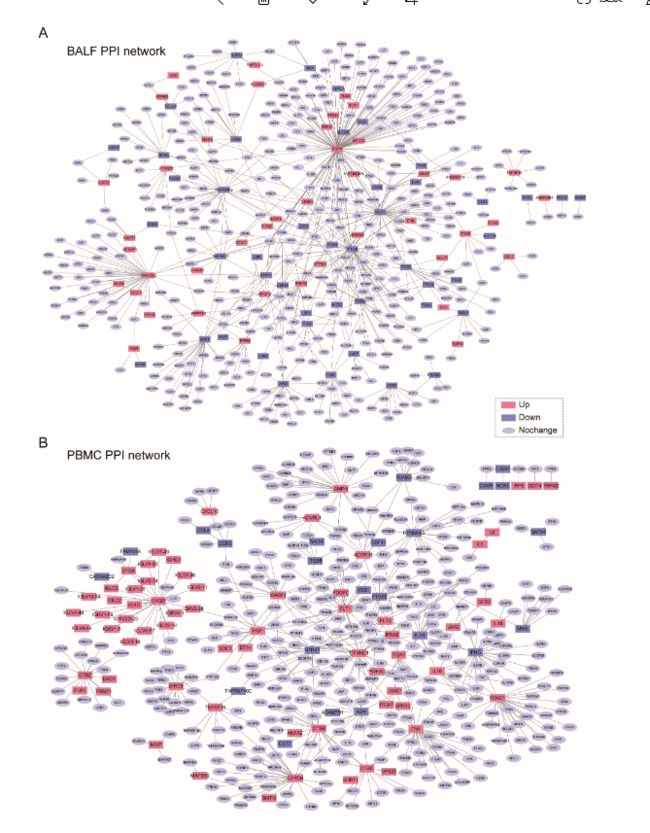
5.Functional enrichment analysis was performed on the list of differentially expressed genes by using the clusterProfiler package (v 3.14.3)to determine if the genes are enriched for specific terms. The level of significance for the enrichment was calculated by a hypergeometric test for each term using all expressed genes as background. The p-values were corrected for multiple hypothesis tests using the Benjamini and Hochberg method to control the false discovery rate (FDR). To inspect genes in specific GO-term and pathway, the GO and KEGG pathway annotation were downloaded from Gene Ontology Resource (Last updated: 22 February 2020) and KEGG database (Last updated: 14 January 2020) , respectively. The interacting proteins to the differentially expressed genes in PBMC or BALF were identified from the functional human network InWeb_InBipMap (a human protein–protein interaction network with direct interaction evidence in high-confidence). Cytoscape (3.5.0) was used for visualizing the PPI subnetwork.
通过使用clusterProfiler程序包(v 3.14.3) 对差异表达的基因列表进行功能富集分析,以确定基因是否针对特定术语富集。
通过超几何学测试,使用所有表达的基因作为背景,计算每个项目的富集显着性水平。
使用Benjamini和Hochberg方法控制错误发现率(FDR),对多个假设检验的p值进行了校正。要检查特定GO术语和途径中的基因,可从Gene Ontology资源(最新更新:2020年2月22日)和KEGG数据库(最新更新:2020年1月14日 下载GO和KEGG途径注释。
PBMC或BALF中与差异表达基因的相互作用蛋白是从功能性的人为网络InWeb_InBipMap(具有高可信度的直接相互作用证据的人蛋白质-蛋白质相互作用网络)中鉴定出来的。Cytoscape(3.5.0)用于可视化PPI子网。
可从https://github.com/zhouyulab/ncov/获得分析的源代码
结果:
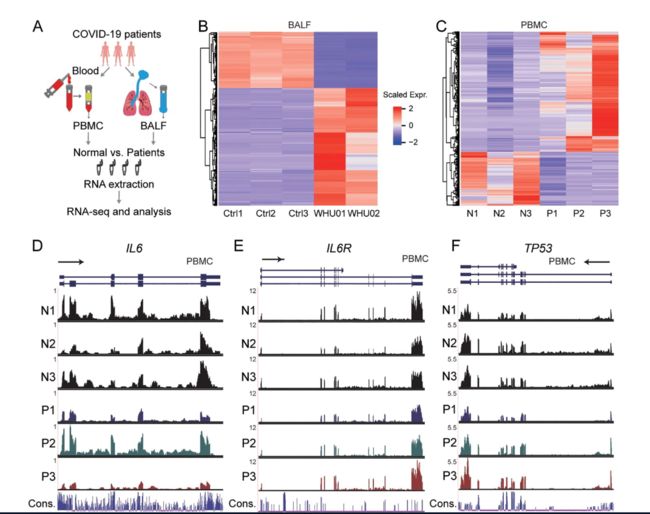
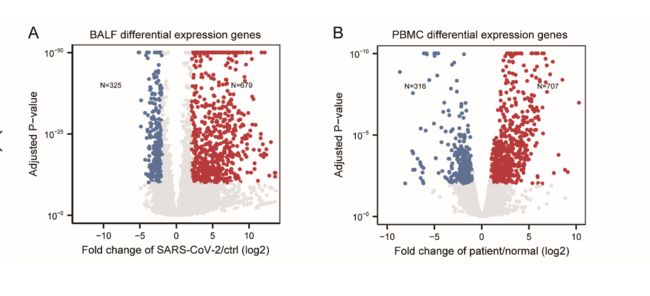
我们对RNA-seq数据进行了质量控制分析,并分别在补充表1和2的BALF和PBMC样本中总结了映射到人类基因组和SARS-CoV-2基因组(GenBank MN988668)的读数的统计信息。
与我们先前针对病毒基因组组装的报告[ 3 ]一致,BALF患者样品包含高比例的病毒读数,而健康对照数据集的计数为零。
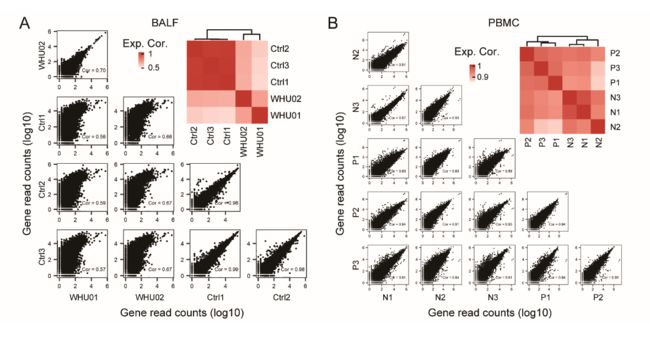
有趣的是,来自患者的PBMC样本几乎没有显示出病毒读数,表明SARS-CoV-2可能不会感染PBMC。数据说明了对照组或患者组之间的高度一致性,分别通过BALF和PBMC样本的相关性和聚类分析得到证明(补充图S1A和S1B)。
有趣的是,如基因组浏览器中显示的RNA-seq信号所示,三分之二的患者IL6和IL6R降低(D,E),尽管TP53表达显示出增加的趋势(F),尽管它们在统计学上没有被确定为显着改变的基因。
调控基因的功能富集分析
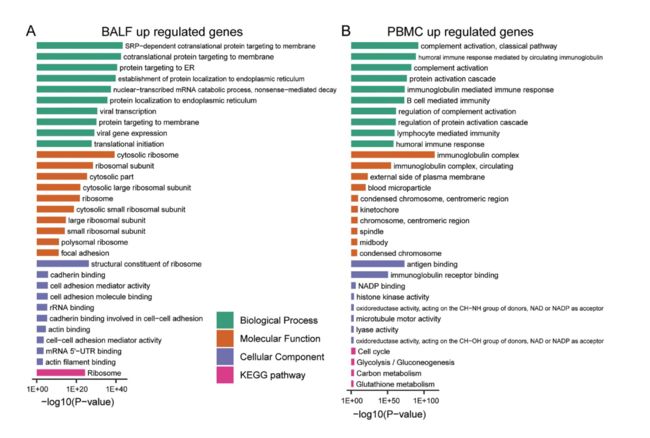
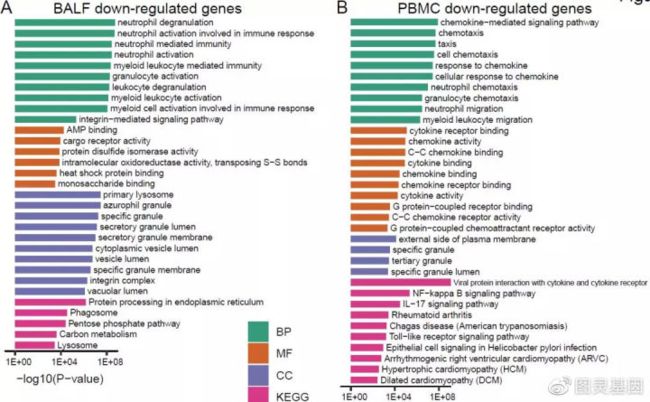
作者对支气管肺泡灌洗液和外周血单核细胞中差异调节基因进行了基因功能富集分析。在支气管肺泡灌洗液样本中,上调的基因与病毒的入侵有关,病毒感染引起各种膜结构和内质网的变化;而在外周血单核细胞样本中上调的基因主要富含“补体激活”、“循环免疫球蛋白介导的体液免疫反应和“ B细胞介导的免疫”,表明外周血单核细胞中的免疫活性已激活;此外,还激活了一系列与炎症相关的过程。
进一步对这些上调和下调的基因进行了KEGG通路分析。在支气管肺泡灌洗液样本中,富集改变基因的途径包括'核糖体','内质网中的蛋白质加工','吞噬体','戊糖磷酸途径','碳代谢'和'溶酶体'。对于外周血单核细胞样本,“细胞周期”和“ p53信号传导”途径丰富了上调的基因,而与细胞因子相关的途径则丰富了下调的基因。
调节细胞因子的分析
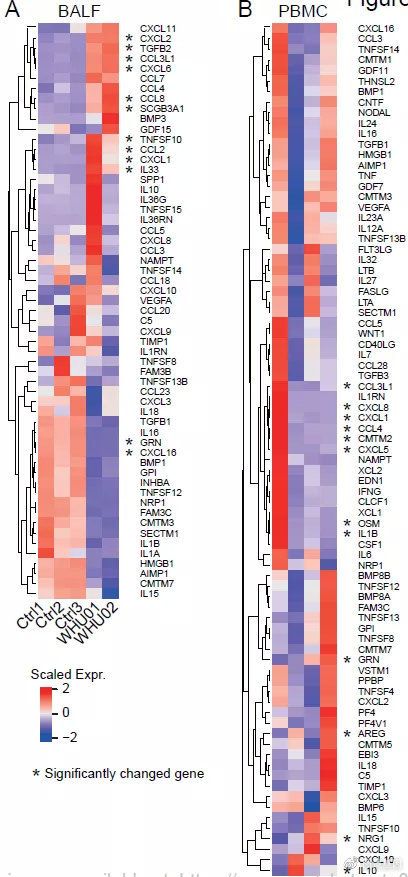
COVID-19患者在SARS-CoV感染会发生细胞因子风暴。我们鉴定了BALF和PBMC中所有表达的细胞因子,其中带有星号标记的基因发生了显着变化。如在临床病例中,在BALF中观察到高细胞因子表达。与实验室检查结果一致,我们发现细胞因子IL10,CCL2 / MCP-1,CXCL10 / IP-10,CCL3 / MIP-1A和CCL4 / MIP1B在患者的BALF样品中高度表达。
两名患者(WHU01和WHU02)之间的进一步比较表明,某些细胞因子的表达在患者之间是可变的,例如IL10,IL36RN,IL36G,TNFSF15,CCL5,TNFSF10,CXCL1和IL33。
SARS-CoV-2感染可能导致淋巴细胞凋亡

3名患者的实验室检查结果(补充表3)表明,包括患者血液中的淋巴细胞在内的各种类型免疫细胞的细胞计数减少。先前的临床和尸检报告表明,患者的淋巴细胞已大大减少。
我们发现一些显着改变的基因丰富了细胞凋亡和P53信号通路([图5],包括CTSL,CTSB,DDIT4,RRAS,CTSD,BIRC5,TNFSF10,CTSZ,NTRK1,IGFBP3,CCNB1,RRM2,CCNB2,GTSE1, CDK1,STEAP3和TP53I3。有趣的是,TP53是凋亡过程中的重要基因,在两名患者中显示出增加的趋势,这表明PMBC的减少可能是由于凋亡引起的。
比较患者BALF和PBMC中不同变化的基因
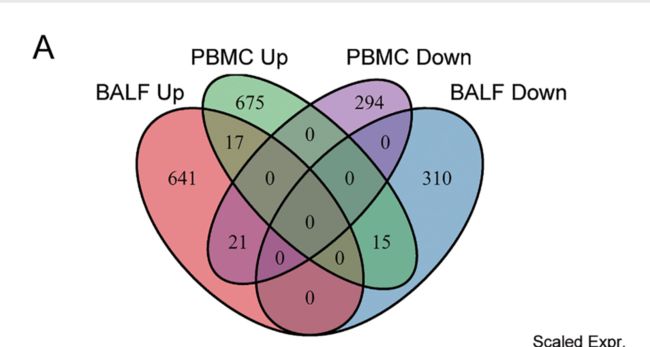
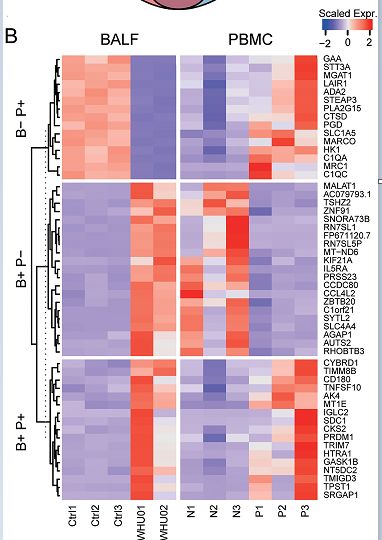
基于差异基因表达分析,我们发现BALF样品和PBMC之间的细胞内基因变化水平存在显着差异。这种区别是由于对两种类型细胞的病毒感染不同。
值得注意的是,该病毒似乎并未感染PBMC,因为我们未能检测到PBMC中的病毒RNA和ACE2表达,这也得到了骨髓血细胞表达数据库的支持。
为了说明两种细胞类型之间的差异,我们对BALF样品和PBMC之间具有不同趋势的基因进行了分类(A,B))。
有17个基因朝同一方向变化,而36个基因则有相反的趋势。
我们首先分析了PBMC中增加但BLAF中减少的基因。在这15个基因中,有5个基因参与了小分子分解代谢过程:ADA2,HK1,MGAT1,PGD和PLA2G15。中性粒细胞的激活和免疫涉及4个基因:ADA2,CTSD,GAA,LAIR1。接下来,我们分析了在BLAF中增加但在PBMC中减少的基因,这些基因在[图6](B)中列出。尽管两个样品之间有17个基因以相同的趋势变化,但生物学意义仍需进一步研究。