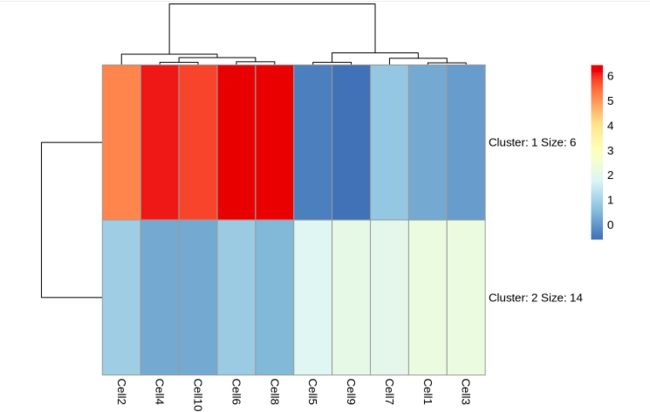
- R可视化之ComplexHeatmap【四】:热图小方格个性化修饰、提取亚集及热图信息
Bio_Infor
特别声明:本部分(系列)内容均来自顾祖光博士对ComplexHeatmap的介绍,仅为学习交流,尊重原创。热图系列我们已经有:R可视化之ComplexHeatmap【一】:颜色、标题、聚类R可视化之ComplexHeatmap【二】:行(列)顺序、行(列)名R可视化之ComplexHeatmap【三】:拆分今天分享:热图小方格个性化修饰、提取亚集及热图信息。热图小方格个性化修饰前段时间很多平台都在
- 使用百度地图绘制热力图(BMapGL)
.ლ.
BMapGL/mapvglvue.js前端
首先要引入百度地图的js、api;(小萌新使用百度地图的时候记得要先申请密钥哦)然后在public下面的index中要先引入相关的js;接下来就可以开始绘制地图啦importBMapGLfrom"BMapGL";import*asmapvglfrom"mapvgl";import{BmlHeatmap}from"vue-baidu-map";//引人exportdefault{components
- pyskl/datasets/pipelines/heatmap_related.py
鱼儿会飞吗
pythonnumpy开发语言
classGeneratePoseTarget:首先看def__call__(self,results):def__call__(self,results):heatmap=self.gen_an_aug(results)key='heatmap_imgs'if'imgs'inresultselse'imgs'ifself.double:indices=np.arange(heatmap.shap
- 11.8 RNA-seq表达rpkm数据操作实践(二)
KK_f2d5
接下来,我们需要对rpkm数据进行annotation再画图。一篇文章说:RPKMvalueswerelog2transformedbeforegeneratingheatmaps.TheheatmapwasgeneratedbyBARHeatmapperPlus(http://bar.utoronto.ca/ntools/cgi-bin/ntools_heatmapper_plus.cgi)on
- R语言绘制热图
小潘的生信笔记
R语言r语言开发语言1024程序员节
运用R语言绘制热图本文主要讲述绘制热图的两种方式,分别为利用pheatmap包和ggplot2包目录运用R语言绘制热图一、热图概念二、热图绘制方法1.利用pheatmap包2.利用ggplot2包一、概念热图是一种很常见的图,其基本原则是用颜色代表数字,让数据呈现更直观、对比更明显。常用来表示不同样品组代表性基因的表达差异、不同样品组代表性化合物的含量差异、不同样品之间的两两相似性。二、绘制方法1
- 利用R中的corrmorant包绘制精美的相关性热图
带我去滑雪
机器学习之Rr语言开发语言
大家好,我是带我去滑雪!相关性热图(correlationheatmap)是一种可视化工具,用于展示数据集中各个变量之间的相关性。它以矩阵的形式显示变量之间的相关系数,并通过色彩编码来表示相关性的强度。在相关性热图中,每个变量都对应图中的一行和一列。图中的每个单元格代表两个变量之间的相关性,通常使用颜色来表示相关性的强度。通常,相关性的计算采用的是Pearson相关系数,它度量线性关系的强度和方向
- R语言画相关性热图
还没秃头的程序猿-D
研究牲r语言开发语言
R语言进行聚类热图install.packages("corrplot2")install.packages("Hmisc")install.packages("pheatmap")library(Hmisc)library("corrplot")library("pheatmap")getwd()#网上随便找的数据集mydata<-mtcars[,c(1:7)]#自己的数据集mydata<-re
- R语言linkET包绘制相关性组合网络热图
天桥下的卖艺者
R语言科研绘图r语言开发语言
热图就是使用颜色来表示数据相关性的图。能绘制热图的R包很多,既往我们已经介绍了《R语言pheatmap包绘制热图》,今天咱们来介绍一下R语言linkET包绘制相关性组合网络热图。相关性热图的意思热图和其他数据的关联性,比较的是多数据间的内在关系。linkET包是我们的一个国人写的,目前需要使用devtools::install_github下载,作者对包的描述是万物皆可互联,还挺有意思的。我们先下
- 分享88个表单按钮JS特效,总有一款适合您
灬沙海灬
javascript
分享88个表单按钮JS特效,总有一款适合您88个表单按钮JS特效下载链接:https://pan.baidu.com/s/1v-qcl8bv2kxZ8a98Xo9UAg?pwd=8888提取码:8888Python采集代码下载链接:采集代码.zip-蓝奏云学习知识费力气,收集整理更不易。知识付费甚欢喜,为咱码农谋福利。heatmap.js-Web动态热图插件jQuery调整表格宽度大小插件(col
- 分享88个表单按钮JS特效,总有一款适合您
自动化新人
javascript
分享88个表单按钮JS特效,总有一款适合您88个表单按钮JS特效下载链接:https://pan.baidu.com/s/1v-qcl8bv2kxZ8a98Xo9UAg?pwd=8888提取码:8888Python采集代码下载链接:采集代码.zip-蓝奏云学习知识费力气,收集整理更不易。知识付费甚欢喜,为咱码农谋福利。heatmap.js-Web动态热图插件jQuery调整表格宽度大小插件(col
- 【R画图】环形热图
jjjscuedu
热图(heatmap)在生信领域基本就是常规操作,基本技能,入门操作。能画热图的工具也有很多,我自己常用的R包是pheatmap。最近经常看见环状的热图,所以就搜了一下资料学习一下,测试一下。环状热图我也经常会在论文中看到,用法和热图相同,但更适合于需要展示较多基因(数据)时来使用。===安装=====install.packages("circlize")install.packages("Co
- pheatmap()画图
多啦A梦的时光机_648d
$rm(list=ls())$library(pheatmap)$a1=rnorm(100)生成一百个随机数的a1$dim(a1)=c(5,20)加维度$pheatmap(a1)$a2=rnorm(100)+2$dim(a2)=c(5,20)$pheatmap(a2)$pheatmap(cbind(a1,a2))$pheatmap(cbind(a1,a2),cluster_cols=T)画的图是自
- 学习:StatQuest-Heatmap
小潤澤
Heatmapimage.png在RNA-seq中热图往往用于衡量不同样本不同基因的表达情况(主要看上下表达),这个图就是个热图,横坐标表示不同样本,纵坐标表示基因。热图中的标准化和聚类Z-scoreimage.png如果有一列数据,我们要计算Z-score:计算这组数据的均值每个数据点减去均值计算标准差用第二步计算的值除以标准差image.png标准化有对某一基因标准化的,有对每个样本进行标准化
- cesium three性能比较_mapboxgl + three 动画 — 网格热图
weixin_39667801
cesiumthree性能比较
一、前言最近工作上需要实现一个地图动画效果,简单的动画效果,是的,简单;不过在此之前,我并不认为它简单,需要有一点点的webgl、three.js的基础。效果大概就是一个网格起伏热力图的动画,像心跳一样、heartbeat……,像这样。Agridheatmap上面这个动效,它好像是基于一个gltf模型来做的,它不给地图交互,现在我们也想实现类似的效果,我们最后实现的效果跟这个效果比还有点差距,我们
- Cesium中三维热力图的实现思路
refineiks
Cesium图形渲染前端
1、普通热力图的绘制基于开源库heatmap.js,将数据组织成它需要的格式heatmap.js需要的输入数据格式2、使用同一份数据通过heatmap.js绘制两张二维热力图,一张正常的热力图,一张用灰度表示的热力图(只有黑白两种颜色的渐变)Heatmap.create({//...gradient:{'0.3':'green','0.5':'blue','0.8':'yellow','0.95'
- 最强Pose模型RTMO开源 | 基于YOLO架构再设计,9MB+9ms性能完爆YOLO-Pose
AI视觉网奇
深度学习基础姿态检测YOLO深度学习
实时多人在图像中的姿态估计面临着在速度和精度之间实现平衡的重大挑战。尽管两阶段的上下文方法在图像中人数增加时会减慢速度,但现有的单阶段方法往往无法同时实现高精度和实时性能。本文介绍了RTMO,这是一个单阶段姿态估计框架,通过在YOLO架构中使用双一维Heatmap来表示关键点,实现与自上而下方法相当的准确度,同时保持高速度。作者提出了一种动态坐标分类器和一种定制的损失函数,用于Heatmap学习,
- org.apache.ibatis.binding.BindingException: Invalid bound statement (not found): com.qingge.springbo
飞滕人生TYF
apache
问题笔者开发项目,使用SpringBoot+Mybatis技术架构,启动控制台报错org.apache.ibatis.binding.BindingException:Invalidboundstatement(notfound):com.qingge.springboot.mapper.WheatMapper.findPage详细问题Mapper层核心代码publicinterfaceWheat
- 日常随笔: React useEffect中使用异步更新数据方法遇到的问题
月半叫做胖
前端学习常见问题ReactReacthooke异步更新useEffect
开发随笔:reacthooks异步更新的问题1.问题叙述在使用antd/G2图表更新的时候发现:在useEffect中通过异步函数对图表渲染的data进行更新,但是由于第一次更新后,数据是延迟更新的,所以好像数据并没有实时进行更新.2.代码分析定义了一个heatmapDataconst[heatMapData,setHeatMapData]=useState([]);异步更新函数//异步更新函数c
- R语言画热图的一个简单小例子2~视频版
小明的数据分析笔记本
大家好,今天继续录制视频介绍R语言的pheatmap包画热图的小例子,今天就是实现下面这幅图B站视频链接https://www.bilibili.com/video/BV1HA411E7gMimage.png
- Pyecharts绘制日历热力图
clearAutumn
python数据可视化数据分析pandas
热力图主要通过颜色去表现数值的大小,必须要配合visualMap组件使用。Pyecharts==0.5.11数据日历热力图所需数据为广州市2018年的空气质量指数数据(guangzhou_AQI_2018.csv),只需要空气质量指数AQI。部分数据截图:代码接下来使用Pandas读取CSV文件,提取数据,Pyecharts绘制日历热力图。frompyechartsimportHeatMapimp
- 存在/缺失矩阵绘制热图
花生学生信
library(pheatmap)#本次所使用的热图绘制包,如果没有安装包,记得提前安装好library(vegan)#vegan包待会可以用来标准化和中心化数据data%mutate(x=1:nrow(.))%>%#select(1:10,x)%>%select(1:ncol(.),x)%>%reshape2::melt(,id.vars="x")%>%mutate(pav=case_when(
- ggplot的热图玩法
小洁忘了怎么分身
背景画热图的体系用的比较多的是pheatmap和ComplexHeatmap这两个包,前者胜在代码简单,功能强大,而后者胜在细节无穷无尽,只有你想不到,没有它做不到。ggplot2在画热图这件事上,是存在感不太强的。但有时候还必须得用它来画,以期和其他ggplot2的图严丝合缝的拼在一起。因此我收集了一下ggplot2的成果,发现又解锁了y叔的一个新包aplot,以及前段时间刚出的ggheatma
- echarts 绘制垂直滚动热力图
Haip
echarts前端javascript
问题1:提示功能无效问题2:值筛选无效效果在线浏览下载echarts官网例子(heatmapExamples-ApacheECharts)稍作改动:generateData入参改为长度和宽度noise.perlin2(i/40,j/20)+Math.random()*5y轴倒置指定zlevel为2通过定时器绘制第一行数据,并将绘制前的图像内容拷贝并下移一个单位constloop=(layers)=
- 第137期 Oracle的数据生命周期管理(20240123)
胖头鱼的鱼缸(尹海文)
Oracle数据库oracle数据库
数据库管理137期2024-01-23第137期Oracle的数据生命周期管理(20240123)1ILM2HeatMap3ADO4优点5对比总结第137期Oracle的数据生命周期管理(20240123)作者:胖头鱼的鱼缸(尹海文)OracleACEAssociate:Database(Oracle与MySQL)网思科技DBA总监10年数据库行业经验,现主要从事数据库服务工作拥有OCM11g/1
- ComplexHeatmap复杂热图绘制学习——4.热图列表
denghb001
热图列表ComplexHeatmap包的主要特点是它可以水平或垂直连接热图和注释列表,我们可以将各种信息来源进行可视化关联。在本章中,我们主要介绍水平串联(在分析中使用频率较高)后面也展示了一些垂直串联的例子。水平和垂直串联在语法上是基本相似的。对于水平串联,所有热图和注释的行数应该相同。我们首先介绍热图的串联,然后我们将展示如何将热图与注释串联。在下面例子中,有三个矩阵,其中第三个是一个热图向量
- openlayers [四] 地图图层 Layers详解
爱玩亚索的程序员
openlayer从入门到进阶javascriptvue.js前端webgis
文章目录1图层1.1线状图层1.2点状图层1.3面状图层2图层存在一个或多个3layers常见的属性4layers各种图层和类型总结在openlayers中,图层是使用layer对象表示的,主要有WebGLPointsLayer、热度图(HeatMapLayer)、图片图层(ImageLayer)、切片图层(TileLayer)和矢量图层(VectorLayer)五种类型,它们都是继承Layer类
- 关于heatmap
果蝇饲养员的生信笔记
一、热图的介绍(1)什么是热图热图是heatmap的直译,用暖色表示数值大,冷色表示数值小。行为基因,列为样本。(2)热图的作用数据质量控制:通过热图,我们能很直观的看到不同分组之间的基因整体表达模式,因此可以迅速地判断同组之间各样本的重复性如何,从而判断实验处理是否正确,数据是否可信可靠符合逻辑。相同实验分组的几个生物学重复应该聚类在一起。直观展示重点研究对象的表达量数据差异变化情况:一次实验中
- heatmap.js + echarts 热力图 折线图渐变色
weixin_54129994
vueVUE+Echartsjavascriptecharts前端
开始停止importHeatmapfrom'heatmap.js'import*asechartsfrom'echarts'exportdefault{name:'Home',data(){return{heatmapInstance:'',timerheat:""}},mounted(){this.show()this.showHeatMap()},methods:{startheat(){th
- echarts地图列表_ECharts系列列表:热力图
韩金虎
echarts地图列表
series[i]-heatmap该节介绍ECharts中用于表示热力图的系列列表。热力图主要通过颜色去表现数值的大小,必须要配合visualMap组件使用。可以应用在直角坐标系以及地理坐标系上,这两个坐标系上的表现形式相差很大,直角坐标系上必须要使用两个类目轴。下面分别是直角坐标系和地理坐标系上应用的例子:直角坐标系:地理坐标系:热力图属性type默认值为heatmapname设置该热力图系列名
- Echarts学习之七:series-heatmap热力图
剑雪封喉r
echarts我的Echarts学习之路
mytextStyle={color:"#333",//文字颜色fontStyle:"normal",//italic斜体oblique倾斜fontWeight:"normal",//文字粗细boldbolderlighter100|200|300|400...fontFamily:"sans-serif",//字体系列fontSize:18,//字体大小};mylineStyle={color:
- 深入浅出Java Annotation(元注解和自定义注解)
Josh_Persistence
Java Annotation元注解自定义注解
一、基本概述
Annontation是Java5开始引入的新特征。中文名称一般叫注解。它提供了一种安全的类似注释的机制,用来将任何的信息或元数据(metadata)与程序元素(类、方法、成员变量等)进行关联。
更通俗的意思是为程序的元素(类、方法、成员变量)加上更直观更明了的说明,这些说明信息是与程序的业务逻辑无关,并且是供指定的工具或
- mysql优化特定类型的查询
annan211
java工作mysql
本节所介绍的查询优化的技巧都是和特定版本相关的,所以对于未来mysql的版本未必适用。
1 优化count查询
对于count这个函数的网上的大部分资料都是错误的或者是理解的都是一知半解的。在做优化之前我们先来看看
真正的count()函数的作用到底是什么。
count()是一个特殊的函数,有两种非常不同的作用,他可以统计某个列值的数量,也可以统计行数。
在统
- MAC下安装多版本JDK和切换几种方式
棋子chessman
jdk
环境:
MAC AIR,OS X 10.10,64位
历史:
过去 Mac 上的 Java 都是由 Apple 自己提供,只支持到 Java 6,并且OS X 10.7 开始系统并不自带(而是可选安装)(原自带的是1.6)。
后来 Apple 加入 OpenJDK 继续支持 Java 6,而 Java 7 将由 Oracle 负责提供。
在终端中输入jav
- javaScript (1)
Array_06
JavaScriptjava浏览器
JavaScript
1、运算符
运算符就是完成操作的一系列符号,它有七类: 赋值运算符(=,+=,-=,*=,/=,%=,<<=,>>=,|=,&=)、算术运算符(+,-,*,/,++,--,%)、比较运算符(>,<,<=,>=,==,===,!=,!==)、逻辑运算符(||,&&,!)、条件运算(?:)、位
- 国内顶级代码分享网站
袁潇含
javajdkoracle.netPHP
现在国内很多开源网站感觉都是为了利益而做的
当然利益是肯定的,否则谁也不会免费的去做网站
&
- Elasticsearch、MongoDB和Hadoop比较
随意而生
mongodbhadoop搜索引擎
IT界在过去几年中出现了一个有趣的现象。很多新的技术出现并立即拥抱了“大数据”。稍微老一点的技术也会将大数据添进自己的特性,避免落大部队太远,我们看到了不同技术之间的边际的模糊化。假如你有诸如Elasticsearch或者Solr这样的搜索引擎,它们存储着JSON文档,MongoDB存着JSON文档,或者一堆JSON文档存放在一个Hadoop集群的HDFS中。你可以使用这三种配
- mac os 系统科研软件总结
张亚雄
mac os
1.1 Microsoft Office for Mac 2011
大客户版,自行搜索。
1.2 Latex (MacTex):
系统环境:https://tug.org/mactex/
&nb
- Maven实战(四)生命周期
AdyZhang
maven
1. 三套生命周期 Maven拥有三套相互独立的生命周期,它们分别为clean,default和site。 每个生命周期包含一些阶段,这些阶段是有顺序的,并且后面的阶段依赖于前面的阶段,用户和Maven最直接的交互方式就是调用这些生命周期阶段。 以clean生命周期为例,它包含的阶段有pre-clean, clean 和 post
- Linux下Jenkins迁移
aijuans
Jenkins
1. 将Jenkins程序目录copy过去 源程序在/export/data/tomcatRoot/ofctest-jenkins.jd.com下面 tar -cvzf jenkins.tar.gz ofctest-jenkins.jd.com &
- request.getInputStream()只能获取一次的问题
ayaoxinchao
requestInputstream
问题:在使用HTTP协议实现应用间接口通信时,服务端读取客户端请求过来的数据,会用到request.getInputStream(),第一次读取的时候可以读取到数据,但是接下来的读取操作都读取不到数据
原因: 1. 一个InputStream对象在被读取完成后,将无法被再次读取,始终返回-1; 2. InputStream并没有实现reset方法(可以重
- 数据库SQL优化大总结之 百万级数据库优化方案
BigBird2012
SQL优化
网上关于SQL优化的教程很多,但是比较杂乱。近日有空整理了一下,写出来跟大家分享一下,其中有错误和不足的地方,还请大家纠正补充。
这篇文章我花费了大量的时间查找资料、修改、排版,希望大家阅读之后,感觉好的话推荐给更多的人,让更多的人看到、纠正以及补充。
1.对查询进行优化,要尽量避免全表扫描,首先应考虑在 where 及 order by 涉及的列上建立索引。
2.应尽量避免在 where
- jsonObject的使用
bijian1013
javajson
在项目中难免会用java处理json格式的数据,因此封装了一个JSONUtil工具类。
JSONUtil.java
package com.bijian.json.study;
import java.util.ArrayList;
import java.util.Date;
import java.util.HashMap;
- [Zookeeper学习笔记之六]Zookeeper源代码分析之Zookeeper.WatchRegistration
bit1129
zookeeper
Zookeeper类是Zookeeper提供给用户访问Zookeeper service的主要API,它包含了如下几个内部类
首先分析它的内部类,从WatchRegistration开始,为指定的znode path注册一个Watcher,
/**
* Register a watcher for a particular p
- 【Scala十三】Scala核心七:部分应用函数
bit1129
scala
何为部分应用函数?
Partially applied function: A function that’s used in an expression and that misses some of its arguments.For instance, if function f has type Int => Int => Int, then f and f(1) are p
- Tomcat Error listenerStart 终极大法
ronin47
tomcat
Tomcat报的错太含糊了,什么错都没报出来,只提示了Error listenerStart。为了调试,我们要获得更详细的日志。可以在WEB-INF/classes目录下新建一个文件叫logging.properties,内容如下
Java代码
handlers = org.apache.juli.FileHandler, java.util.logging.ConsoleHa
- 不用加减符号实现加减法
BrokenDreams
实现
今天有群友发了一个问题,要求不用加减符号(包括负号)来实现加减法。
分析一下,先看最简单的情况,假设1+1,按二进制算的话结果是10,可以看到从右往左的第一位变为0,第二位由于进位变为1。
- 读《研磨设计模式》-代码笔记-状态模式-State
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
/*
当一个对象的内在状态改变时允许改变其行为,这个对象看起来像是改变了其类
状态模式主要解决的是当控制一个对象状态的条件表达式过于复杂时的情况
把状态的判断逻辑转移到表示不同状态的一系列类中,可以把复杂的判断逻辑简化
如果在
- CUDA程序block和thread超出硬件允许值时的异常
cherishLC
CUDA
调用CUDA的核函数时指定block 和 thread大小,该大小可以是dim3类型的(三维数组),只用一维时可以是usigned int型的。
以下程序验证了当block或thread大小超出硬件允许值时会产生异常!!!GPU根本不会执行运算!!!
所以验证结果的正确性很重要!!!
在VS中创建CUDA项目会有一个模板,里面有更详细的状态验证。
以下程序在K5000GPU上跑的。
- 诡异的超长时间GC问题定位
chenchao051
jvmcmsGChbaseswap
HBase的GC策略采用PawNew+CMS, 这是大众化的配置,ParNew经常会出现停顿时间特别长的情况,有时候甚至长到令人发指的地步,例如请看如下日志:
2012-10-17T05:54:54.293+0800: 739594.224: [GC 739606.508: [ParNew: 996800K->110720K(996800K), 178.8826900 secs] 3700
- maven环境快速搭建
daizj
安装mavne环境配置
一 下载maven
安装maven之前,要先安装jdk及配置JAVA_HOME环境变量。这个安装和配置java环境不用多说。
maven下载地址:http://maven.apache.org/download.html,目前最新的是这个apache-maven-3.2.5-bin.zip,然后解压在任意位置,最好地址中不要带中文字符,这个做java 的都知道,地址中出现中文会出现很多
- PHP网站安全,避免PHP网站受到攻击的方法
dcj3sjt126com
PHP
对于PHP网站安全主要存在这样几种攻击方式:1、命令注入(Command Injection)2、eval注入(Eval Injection)3、客户端脚本攻击(Script Insertion)4、跨网站脚本攻击(Cross Site Scripting, XSS)5、SQL注入攻击(SQL injection)6、跨网站请求伪造攻击(Cross Site Request Forgerie
- yii中给CGridView设置默认的排序根据时间倒序的方法
dcj3sjt126com
GridView
public function searchWithRelated() {
$criteria = new CDbCriteria;
$criteria->together = true; //without th
- Java集合对象和数组对象的转换
dyy_gusi
java集合
在开发中,我们经常需要将集合对象(List,Set)转换为数组对象,或者将数组对象转换为集合对象。Java提供了相互转换的工具,但是我们使用的时候需要注意,不能乱用滥用。
1、数组对象转换为集合对象
最暴力的方式是new一个集合对象,然后遍历数组,依次将数组中的元素放入到新的集合中,但是这样做显然过
- nginx同一主机部署多个应用
geeksun
nginx
近日有一需求,需要在一台主机上用nginx部署2个php应用,分别是wordpress和wiki,探索了半天,终于部署好了,下面把过程记录下来。
1. 在nginx下创建vhosts目录,用以放置vhost文件。
mkdir vhosts
2. 修改nginx.conf的配置, 在http节点增加下面内容设置,用来包含vhosts里的配置文件
#
- ubuntu添加admin权限的用户账号
hongtoushizi
ubuntuuseradd
ubuntu创建账号的方式通常用到两种:useradd 和adduser . 本人尝试了useradd方法,步骤如下:
1:useradd
使用useradd时,如果后面不加任何参数的话,如:sudo useradd sysadm 创建出来的用户将是默认的三无用户:无home directory ,无密码,无系统shell。
顾应该如下操作:
- 第五章 常用Lua开发库2-JSON库、编码转换、字符串处理
jinnianshilongnian
nginxlua
JSON库
在进行数据传输时JSON格式目前应用广泛,因此从Lua对象与JSON字符串之间相互转换是一个非常常见的功能;目前Lua也有几个JSON库,本人用过cjson、dkjson。其中cjson的语法严格(比如unicode \u0020\u7eaf),要求符合规范否则会解析失败(如\u002),而dkjson相对宽松,当然也可以通过修改cjson的源码来完成
- Spring定时器配置的两种实现方式OpenSymphony Quartz和java Timer详解
yaerfeng1989
timerquartz定时器
原创整理不易,转载请注明出处:Spring定时器配置的两种实现方式OpenSymphony Quartz和java Timer详解
代码下载地址:http://www.zuidaima.com/share/1772648445103104.htm
有两种流行Spring定时器配置:Java的Timer类和OpenSymphony的Quartz。
1.Java Timer定时
首先继承jav
- Linux下df与du两个命令的差别?
pda158
linux
一、df显示文件系统的使用情况,与du比較,就是更全盘化。 最经常使用的就是 df -T,显示文件系统的使用情况并显示文件系统的类型。 举比例如以下: [root@localhost ~]# df -T Filesystem Type &n
- [转]SQLite的工具类 ---- 通过反射把Cursor封装到VO对象
ctfzh
VOandroidsqlite反射Cursor
在写DAO层时,觉得从Cursor里一个一个的取出字段值再装到VO(值对象)里太麻烦了,就写了一个工具类,用到了反射,可以把查询记录的值装到对应的VO里,也可以生成该VO的List。
使用时需要注意:
考虑到Android的性能问题,VO没有使用Setter和Getter,而是直接用public的属性。
表中的字段名需要和VO的属性名一样,要是不一样就得在查询的SQL中
- 该学习笔记用到的Employee表
vipbooks
oraclesql工作
这是我在学习Oracle是用到的Employee表,在该笔记中用到的就是这张表,大家可以用它来学习和练习。
drop table Employee;
-- 员工信息表
create table Employee(
-- 员工编号
EmpNo number(3) primary key,
-- 姓