from 生信技能树
paper: Brookes, E. et al. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell 10, 157–170 (2012).
GSE accession: GSE34518,总共是9个样本,但是很多样本都分开在多个lane测序的,所以每个样本其实是有多个sra文件,需要进行合并。
Use prefetch to download them all, then transform those SRA files to fastq files by sra-toolkits, then align them to mm10, and call peaks.
测序用的是: Illumina Genome Analyzer II 测序仪,测序策略是 SE50。
data="/home/zzz/data/test"
#查找sra accesion list,用vim命令写入sra.list文件
SRR391032
SRR391033
SRR391034
SRR391035
SRR391036
SRR391037
SRR391038
SRR391039
SRR391040
SRR391041
SRR391042
SRR391043
SRR391044
SRR391045
SRR391046
SRR391047
SRR391048
SRR391049
SRR391050
下载sra并且转换为fastq
cd $data
conda install -c bioconda sra-tools #安装sra-tools
cd $data/sra
cat sra.list |while read i; do prefetch $i; done #download sra files
## 默认下载目录:~/ncbi/public/sra/ ,但是我的是下载在当前文件夹下的,而且每个sra文件都是一个文件夹。
ls -lh ~/ncbi/public/sra/
第一步需要制作配置文件,需要先从ncbi下载sra的information文件sra.table
## 直接用excel制作config文件,或者写代码, 我当然是用的excel,这个代码太麻烦了啦。。。
cut -f 4,7 sra.table |cut -d":" -f 2 |sed 's/ChIPSeq//g' | sed 's/MockIP//g'|sed 's/^ //' |tr ' ' '_' |perl -alne '{$h{$F[0]}++ if exists $h{$F[0]}; $h{$F[0]}=1 unless exists $h{$F[0]};print "$F[0]$h{$F[0]}\t$F[1]"}' > config
得到内容如下,命名为config.txt
RNAPII_S5P_1 SRR391032
RNAPII_S5P_2 SRR391033
RNAPII_S2P_1 SRR391034
RNAPII_S7P_1 SRR391035
RNAPII_8WG16_1 SRR391036
RNAPII_8WG16_2 SRR391037
RNAPII_S2P_2 SRR391038
RNAPII_S2P_3 SRR391039
RNAPII_S7P_2 SRR391040
H2Aub1_1 SRR391041
H2Aub1_2 SRR391042
H3K36me3_1 SRR391043
H3K36me3_2 SRR391044
Control_1 SRR391045
Control_2 SRR391046
Ring1B_1 SRR391047
Ring1B_2 SRR391048
Ring1B_3 SRR391049
RNAPII_S5PRepeat_1 SRR391050
批量sra转fq文件
## 下面用到的 config 文件,就是上面自行制作的。
cat config.txt |while read i;
do echo $i
arr=($i)
srr=${arr[1]}
sample=${arr[0]}
# 单端测序数据的sra转fasq
fastq-dump -A $sample -O $data/raw --gzip --split-3 $data/sra; done
# -A: Replaces accession derived from in filename(s) and deflines (only for single table dump)
#不知道为什么我的这个命令就是跑不通,所以我用了自己的代码:
fastq-dump --gzip --split-3 $data/sra/*sra
#然后把fastq.gz文件手动改了名称,其实可以下载的时候直接用-O参数重新命名。
qc
cd $data/raw/
ls *gz | xargs fastqc -t 10 -o $data/raw/qc
multiqc $data/raw/qc -o $data/raw/qc
用trim_galore进行数据过滤,此处是单端测序
cd $data/raw/
ls *gz | while read i;
do
nohup trim_galore -q 25 --phred33 --length 25 -e 0.1 --stringency 4 -o $data/clean $i &
done
trimmed_data进行QC
mkdir $data/clean/qc
ls $data/clean/*gz | xargs fastqc -t 10 -o $data/clean/qc
multiqc $data/clean/qc -o $data/raw/qc
使用bowtie2进行比对
直接用bowtie2进行比对和统计比对率, 需要提前下载参考基因组然后使用命令构建索引,或者直接就下载索引文件,这里用常用的mm10
# 索引大小为3.2GB, 不建议自己下载基因组构建,可以直接下载索引文件,代码如下:
mkdir referece && cd reference
wget -4 -q ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/mm10.zip
unzip mm10.zip
单端测序数据进行比对
ls ../clean/*gz |while read i;
do
file=$(basename $i)
sample=${file%%.*}
# echo $file $sample
bowtie2 -p 5 -x $bowtie2_index -U $i | samtools sort -O bam -@ 5 -o - > ${sample}.bam
done #最后只输出bam文件,没有sam文件
注意到这里没有用samtools 的view命令将sam转换成bam文件,然后再sort,我就比较了一下这两个命令的区别。下面是结果:
Activated-24h-1.bam.count.txt:ENSMUSG00000000028 3092
Activated-24h-2.bam.count.txt:ENSMUSG00000000028 3483
Activated-72h-1.bam.count.txt:ENSMUSG00000000028 2260
Activated-72h-2.bam.count.txt:ENSMUSG00000000028 1989
Resting-1.bam.count.txt:ENSMUSG00000000028 157
Resting-2.bam.count.txt:ENSMUSG00000000028 185
对过滤后mapping得到的bam文件进行QC
cd $data/bam
ls *.bam |xargs -i samtools index {}
ls *.bam |while read i ;do (nohup samtools flagstat $i > $(basename $i ".bam").stat & );done
grep '%' *stat #得到所有mapping率
Control_1_trimmed.stat:7415799 + 0 mapped (87.76% : N/A)
Control_2_trimmed.stat:7202987 + 0 mapped (86.18% : N/A)
H2Aub1_1_trimmed.stat:8949983 + 0 mapped (97.19% : N/A)
H2Aub1_2_trimmed.stat:13195346 + 0 mapped (97.27% : N/A)
H3K36me3_1_trimmed.stat:11732645 + 0 mapped (98.85% : N/A)
H3K36me3_2_trimmed.stat:13404798 + 0 mapped (98.38% : N/A)
Ring1B_1_trimmed.stat:4620324 + 0 mapped (93.31% : N/A)
Ring1B_2_trimmed.stat:4633085 + 0 mapped (93.57% : N/A)
Ring1B_3_trimmed.stat:22884650 + 0 mapped (95.12% : N/A)
RNAPII_8WG16_1_trimmed.stat:7465754 + 0 mapped (96.07% : N/A)
RNAPII_8WG16_2_trimmed.stat:20652996 + 0 mapped (95.30% : N/A)
RNAPII_S2P_1_trimmed.stat:24966425 + 0 mapped (97.05% : N/A)
RNAPII_S2P_2_trimmed.stat:6095629 + 0 mapped (94.73% : N/A)
RNAPII_S2P_3_trimmed.stat:8659690 + 0 mapped (96.81% : N/A)
RNAPII_S5P_1_trimmed.stat:11791542 + 0 mapped (97.45% : N/A)
RNAPII_S5P_2_trimmed.stat:12171421 + 0 mapped (98.09% : N/A)
RNAPII_S5PRepeat_1_trimmed.stat:4158664 + 0 mapped (82.71% : N/A)
RNAPII_S7P_1_trimmed.stat:6378735 + 0 mapped (80.81% : N/A)
RNAPII_S7P_2_trimmed.stat:5962539 + 0 mapped (82.54% : N/A)
有几个文件建index的时候报错,查了网站之后,用了下面的命令,发现正常的bam文件返回值是VALID,而报错的文件不是。因此我重新跑了QC命令之后就好了。
gunzip -t H2Aub1_1_trimmed.bam && echo "VALID"
gzip: H2Aub1_1_trimmed.bam: decompression OK, trailing garbage ignored
gunzip -t Control_1_trimmed.bam && echo "VALID"
VALID
合并bam文件
samtools 的merge命令只能用来merge sort过的bam文件
当有多个bam文件时,一般思路就是对每一个bam进行sort、index后,再merge成一个整体merged.bam,然后对merged.bam再进行sort、index,才算能用了,得到最终结果应该是是sorted.merge.bam
## 如果不用循环:
## samtools merge control.merge.bam Control_1_trimmed.bam Control_2_trimmed.bam
## 循环命令
mkdir ~/project/epi/ mergeBam
cd ~/project/epi/align
ls *.bam|sed 's/_[0-9]_trimmed.bam//g' |sort -u |while read id;do samtools merge $data/merge.bam/$i.merge.bam $i*.bam ;done
#sort -u 的意思是输出的时候去除重复
mapping过的bam文件,进行筛选,qc
#去除duplicate
ls $data/merge.bam/*merge.bam | while read i ;do (nohup samtools markdup -r $i $(basename $id ".bam").rmdup.bam & );done #这样写nohup命令虽然可以几个同时跑,比较快,但是我不知道nohup命令的log文件被写到哪里了。
#去除低mapping的reads,以及mapping多次的reads
ls $data/merge.bam/*rmdup.bam | while read i;
do
file=$(basename $i)
sample=${file%%.*}
echo $file $sample
samtools view $i -bF 4 -q 10 > ${sample}.uniq.bam
done
#过滤后的reads进行qc
ls $data/merge.bam/*.uniq.bam |xargs -i samtools index {}
ls $data/merge.bam/*.uniq.bam |while read i ;do
samtools flagstat $i > $(basename $i ".uniq.bam").stat;done
grep '%' *stat
control.stat:10120184 + 0 mapped (100.00% : N/A)
H2Aub1.stat:14453515 + 0 mapped (100.00% : N/A)
H3K36me3.stat:20416595 + 0 mapped (100.00% : N/A)
Ring1B.stat:20587109 + 0 mapped (100.00% : N/A)
RNAPII_8WG16.stat:19619979 + 0 mapped (100.00% : N/A)
RNAPII_S2P.stat:21815365 + 0 mapped (100.00% : N/A)
RNAPII_S5PRepeat.stat:3324476 + 0 mapped (100.00% : N/A)
RNAPII_S5P.stat:7593988 + 0 mapped (100.00% : N/A)
RNAPII_S7P.stat:8086221 + 0 mapped (100.00% : N/A)
使用macs2进行call peak
cd $data/merge.bam
ls *uniq.bam |cut -d"." -f 1 |while read i;
do
macs2 callpeak -c control.uniq.bam -t $i.uniq.bam -f BAM -B -g mm -n $i --outdir $data/peaks 2 > $id.log
done
-t: 实验组的输出结果
-c: 对照组的输出结果
-f: -t和-c提供文件的格式,可以是”ELAND”, “BED”, “ELANDMULTI”, “ELANDEXPORT”, “ELANDMULTIPET” (for pair-end tags), “SAM”, “BAM”, “BOWTIE”, “BAMPE” “BEDPE” 任意一个。如果不提供这项,就是自动检测选择。
-g: 基因组大小, 默认提供了hs, mm, ce, dm选项, 不在其中的话,比如说拟南芥,就需要自己提供了。
-n: 输出文件的前缀名
-B: 会保存更多的信息在bedGraph文件中,如fragment pileup, control lambda, -log10pvalue and -log10qvalue scores, 但这个参数意义不大,得到的bedgraph文件没啥用。
-q: q值,也就是最小的PDR阈值, 默认是0.05。q值是根据p值利用BH计算,也就是多重试验矫正后的结果。
-p: 这个是p值,指定p值后MACS2就不会用q值了。
-m: 和MFOLD有关,而MFOLD和MACS预构建模型有关,默认是5:50,MACS会先寻找100多个peak区构建模型,一般不用改,因为你很大概率上不会懂。
对比了一下只去掉adaptor,去掉adaptor和duplicate,去除adaptor以及没有比对上、多重比对、duplicate的peaks(raw_peaks,rmdup_peaks,peaks)
wc -l *bed
#raw_peaks
0 ../raw_peaks/control_summits.bed
1182 ../raw_peaks/H2Aub1_summits.bed
40034 ../raw_peaks/H3K36me3_summits.bed
26029 ../raw_peaks/Ring1B_summits.bed
41628 ../raw_peaks/RNAPII_8WG16_summits.bed
20029 ../raw_peaks/RNAPII_S2P_summits.bed
38659 ../raw_peaks/RNAPII_S5PRepeat_summits.bed
56672 ../raw_peaks/RNAPII_S5P_summits.bed
72203 ../raw_peaks/RNAPII_S7P_summits.bed
296436 total
#rmdup_peaks
0 control_summits.bed
1182 H2Aub1_summits.bed
39815 H3K36me3_summits.bed
26029 Ring1B_summits.bed
41628 RNAPII_8WG16_summits.bed
20029 RNAPII_S2P_summits.bed
38659 RNAPII_S5PRepeat_summits.bed
56750 RNAPII_S5P_summits.bed
72203 RNAPII_S7P_summits.bed
296295 total
#peaks
0 ../peaks/control_summits.bed
2203 ../peaks/H2Aub1_summits.bed
23031 ../peaks/H3K36me3_summits.bed
25062 ../peaks/Ring1B_summits.bed
34926 ../peaks/RNAPII_8WG16_summits.bed
20118 ../peaks/RNAPII_S2P_summits.bed
33668 ../peaks/RNAPII_S5PRepeat_summits.bed
54452 ../peaks/RNAPII_S5P_summits.bed
62553 ../peaks/RNAPII_S7P_summits.bed
256013 total
发现去不去PCR duplicate对于最后call peaks的结果不会影响太大,但是去不去多重比对和没有比对对结果影响比较大。
根据生信技能树的经验:
前几个月处理这个数据集的时候使用的过滤低质量reads参数是短于 35bp的全部丢弃,现在是短于25bp的全部抛弃,导致了得到的peaks从数量上千差别不小。
MACS2结果
_peaks.xls
This file is generated by MACS version 2.2.6
Command line: callpeak -c Activated_input.bam -t Activated_H2AK9ac.bam -f BAM -B -g mm -n Activated_H2AK9ac
ARGUMENTS LIST:
name = Activated_H2AK9ac
format = BAM
ChIP-seq file = ['Activated_H2AK9ac.bam']
control file = ['Activated_input.bam']
effective genome size = 1.87e+09
band width = 300
model fold = [5, 50]
qvalue cutoff = 5.00e-02
The maximum gap between significant sites is assigned as the read length/tag size.
The minimum length of peaks is assigned as the predicted fragment length "d".
Larger dataset will be scaled towards smaller dataset.
Range for calculating regional lambda is: 1000 bps and 10000 bps
Broad region calling is off
Paired-End mode is off
tag size is determined as 50 bps
total tags in treatment: 25164589
tags after filtering in treatment: 25164589
maximum duplicate tags at the same position in treatment = 1
Redundant rate in treatment: 0.00
total tags in control: 18816543
tags after filtering in control: 18816543
maximum duplicate tags at the same position in control = 1
Redundant rate in control: 0.00
d = 214
alternative fragment length(s) may be 214 bps
chr start end length abs_summit pileup -log10(pvalue) fold_enrichment -log10(qvalue) name
chr1 4784036 4784310 275 4784120 17.20 8.44714 4.54950 6.37316 Activated_H2AK9ac_peak_1
#length: peak区域长度
#abs_summit: peak的峰值位点(summit position)
#pileup: peak 峰值的高度(pileup height at peak summit, -log10(pvalue) for the peak summit)
#fold_enrichment: peak的富集倍数(相对于random Poisson distribution with local lambda)
Coordinates in xls is 1-based which is different with BED format
xls里的坐标和bed格式的坐标还不一样,起始坐标需要减1才与narrowPeak的起始坐标一样。
_peaks.narrowPeak
narrowPeak文件是BED6+4格式,可以上传到UCSC浏览。输出文件每列信息分别包含:
1;染色体号
2:peak起始位点
3:结束位点
4:peak name
5:int(-10*log10qvalue)
6 :正负链
7:fold change
8:-log10pvalue
9:-log10qvalue
10:relative summit position to peak start(?)
chr1 4784035 4784310 Activated_H2AK9ac_peak_1 63 . 4.54950 8.44714 6.37316 84
chr1 4784829 4785734 Activated_H2AK9ac_peak_2 586 . 12.83651 61.82663 58.671
_summits.bed
BED格式的文件,包含peak的summits位置,第5列是-log10pvalue。如果想找motif,推荐使用此文件。
chr1 3670687 3670688 Activated_H3K27me3_peak_1 7.21477
chr1 3671910 3671911 Activated_H3K27me3_peak_2 16.00848
补充:
bed格式简介
BED格式能够非常简洁的表示基因组特征和注释,尽管BED格式描述中定义了12列,但是仅仅只有3列必须,因此BED格式按照列数继续细分为BED3,BED4,BED5,BED6,BED12。
BED12定义的12列分别为:chrom, start, end, name(BED代表的特征名),score(范围为0~1000,可以是pvalue, 或者是字符串,如"up"), strand(正负链), thickstart, thickednd(额外着色位置, 比如说表示外显子), itemRgb(RGB颜色,如255,0,0), blockCount(区块数量, 如外显子), blockSizes(由逗号隔开的区块大小), blockStarts(由逗号隔开的区块起始位点)。
对BED文件的细分格式进行举例说明
BED3:chr1 11873 14409
BED4: chr1 11873 14409 uc001aaa.3
BED5: chr1 11873 14409 uc001aaa.3 0
BED6: chr1 11873 14409 uc001aaa.3 0 +
BED12: chr1 11873 14409 uc001aaa.3 0 + 11873 12000 123,123,123 3 354,109,1189, 0,739,1347,
1-based coordinate system:序列的第一个碱基设为数字1,如SAM, VCF, GFF, wiggle格式
0-based coordinate system :序列的第一个碱基设为数字0,如BAM, BCFv2, BED, PSL格式
.bdg
bedGraph格式,可以导入UCSC或者转换为bigwig格式。两种bfg文件:treat_pileup, and control_lambda.
NAME_peaks.broadPeak
BED6+3格式与narrowPeak类似,只是没有第10列。
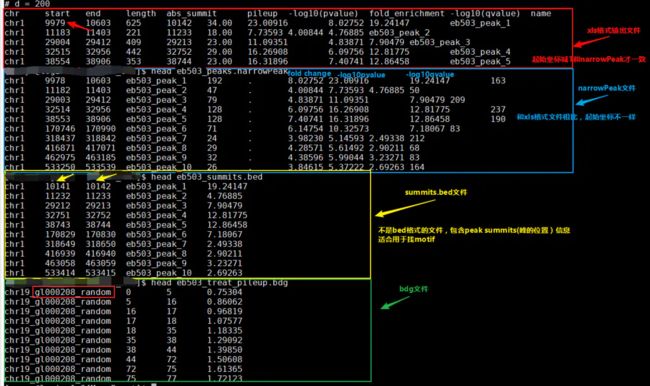
上图引自: 用MACS2软件call peaks
使用deeptools进行可视化
还有很多其他用法,参考:https://vip.biotrainee.com/d/226-如何使用deeptools处理bam数据
deeptools提供bamCoverage和bamCompare进行格式转换,为了能够比较不同的样本,需要先将基因组分成等宽分箱(bin),统计每个分箱的read数,最后得到描述性统计值。对于两个样本,描述性统计值可以是两个样本的比率,或是比率的log2值,或者是差值。如果是单个样本,可以用SES方法进行标准化。
bamCoverage的基本用法
bamCoverage -e 170 -bs 10 -b chip_sorted.bam -o chip.bw
# chip_sorted.bam是前期比对得到的BAM文件
得到的bw文件就可以送去IGV/Jbrowse进行可视化。 这里的参数仅使用了-e/--extendReads和-bs/--binSize即拓展了原来的read长度,且设置分箱的大小。其他参数还有
--filterRNAstrand {forward, reverse}: 仅统计指定正链或负链
--region/-r CHR:START:END:选取某个区域统计
--smoothLength:通过使用分箱附近的read对分箱进行平滑化
如果为了其他结果进行比较,还需要进行标准化,deeptools提供了如下参数:
--scaleFactor:缩放系数
--normalizeUsingRPKMReads: Per Kilobase per Million mapped reads (RPKM)标准化
--normalizeTo1x: 按照1x测序深度(reads per genome coverage, RPGC)进行标准化
--ignoreForNormalization: 指定那些染色体不需要经过标准化
如果需要以100为分箱,并且标准化到1x,且仅统计某一条染色体区域的正链,输出格式为bedgraph,那么命令行可以这样写
mouse测序数据,2号染色体中间会浪费一堆reads
bedgraph转换成tdf格式,igvtools可以完成转换,tdf格式文件可以快速被igv打开
查看TSS附近的信号强度
#先得到bw文件,之前已经得到,这里就不再赘述
bed="/home/zzz/data/reference/annotation/mm10_refseq.bed"
for i in $data/bw/*rmdup.bw;
do
echo $i
file=$(basename $i )
sample=${file%%.*}
echo $sample
computeMatrix reference-point --referencePoint TSS \
-b 2000 -a 2000 \
-R $bed \
-S $i \
--skipZeros -o matrix1_${sample}_TSS_2K.gz \
--outFileSortedRegions ${sample}_TSS_2K.bed
# 输出的gz为文件用于plotHeatmap, plotProfile
## both plotHeatmap and plotProfile will use the output from computeMatrix
plotHeatmap -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Heatmap_2K.png
plotHeatmap -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Heatmap_2K.pdf --plotFileFormat pdf --dpi 720
plotProfile -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Profile_2K.png
plotProfile -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Profile_2K.pdf --plotFileFormat pdf --perGroup --dpi 720
done
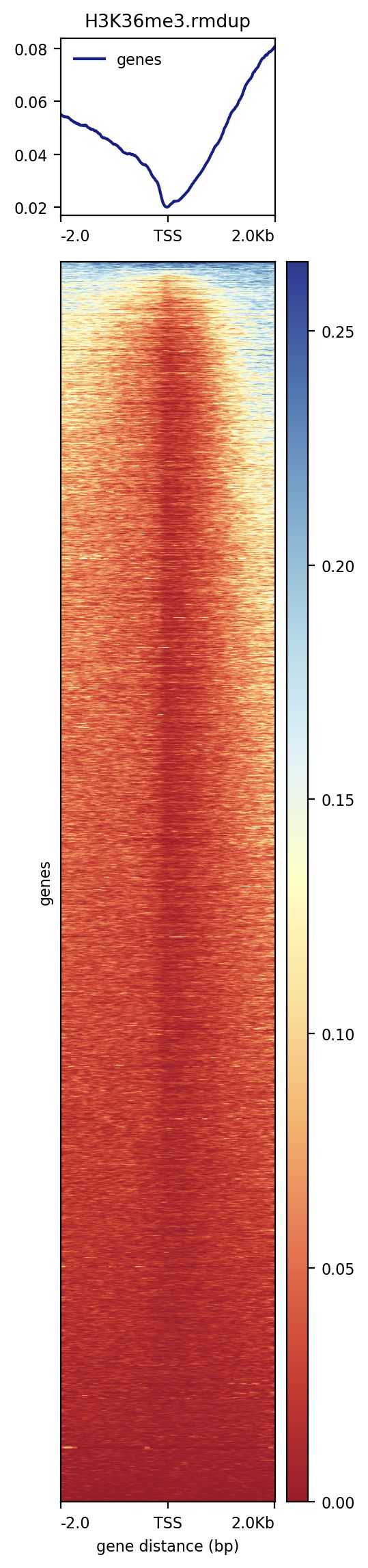
画genebody的图
bed="/home/zzz/data/reference/annotation/mm10_refseq.bed"
for i in $data/bw/*.rmdup.bw ;
do
echo $i
file=$(basename $i )
sample=${file%%.*}
echo $sample
computeMatrix scale-regions -p 5 \
-b 3000 -a 3000 \
-R $bed \
-S $i \
--regionBodyLength 15000 --skipZeros \
-o matrix1_${sample}_genebody.gz \
--outFileSortedRegions regions1_${sample}_genebody.bed
## both plotHeatmap and plotProfile will use the output from computeMatrix
plotHeatmap -m matrix1_${sample}_genebody.gz -out ${sample}_Heatmap_genebody.png
plotHeatmap -m matrix1_${sample}_genebody.gz -out ${sample}_Heatmap_genebody.pdf --plotFileFormat pdf --dpi 720 #输出文件有两种格式,一个是png,一个是pdf,实操的时候可以只保留一个。
plotProfile -m matrix1_${sample}_genebody.gz -out ${sample}_Profile_genebody.png
plotProfile -m matrix1_${sample}_genebody.gz -out ${sample}_Profile_genebody.pdf --plotFileFormat pdf --perGroup --dpi 720
done
上面的批量代码其实就是为了统计全基因组范围的peak在基因特征的分布情况,也就是需要用到computeMatrix计算,用plotHeatmap以热图的方式对覆盖进行可视化,用plotProfile以折线图的方式展示覆盖情况。
computeMatrix具有两个模式:scale-region和reference-point。前者用来信号在一个区域内分布,后者查看信号相对于某一个点的分布情况。无论是那个模式,都有两个参数是必须的,-S是提供bigwig文件,-R是提供基因的注释信息。还有更多个性化的可视化选项。
使用R包对找到的peaks文件进行注释
bedPeaksFile ='8WG16_summits.bed';
bedPeaksFile
## loading packages
require(ChIPseeker)
require(TxDb.Mmusculus.UCSC.mm10.knownGene)
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
require(clusterProfiler)
peak <- readPeakFile( bedPeaksFile )
keepChr= !grepl('_',seqlevels(peak)) #将chr_去掉,只留下常染色体和性染色体
seqlevels(peak, pruning.mode="coarse") <- seqlevels(peak)[keepChr]
peakAnno <- annotatePeak(peak, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Mm.eg.db")
peakAnno_df <- as.data.frame(peakAnno)
更详细的教程参见另一篇分享:https://www.jianshu.com/p/a4f497608485
homer
homer安装
conda install homer
homer 软件配置
mkdir ~software/homer && cd homer
wget http://homer.ucsd.edu/homer/configureHomer.pl
# conda安装的Homer实际没有包含参考序列或注释数据 ;但是可以使用 configureHomer.pl下载数据
perl /home/zzz/data/software/homer/configureHomer.pl -install mm10
Usage: configureHomer.pl [options]
Options:
-list 列出可用的包
-install 安装Homer或homer需要用到的数据包
-version 安装homer包时,可以指定包版本
-remove 移除包
-update 更新所有包到最新版本
-reinstall 强制重新安装所有已经安装过的包
-all 安装所有包
-getFacts (add humor to HOMER - to remove delete contents of data/misc/)
-check 检查第三方软件:samtools, DESeq2, edgeR
-make 重新配置和编译可执行文件
-sun SunOS系统,使用gmake 和 gtar代替make 和 tar
-keepScript 不更新configureHomer.pl
-url 安装时,使用的资源地址,默认:http://homer.ucsd.edu/homer/
Hubs & BigWig settings (with read existing settings from config.txt if upgrading):
-bigWigUrl homer 使用
cd $data/peaks
for i in *.bed;
do
echo $i
file=$(basename $i )
sample=${file%%.*}
echo $sample
awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' $i >${sample}.bed ## 将bed文件的第4列,第1列,第2列,第3列打印出来,是为了让bed文件符合homer软件的输入
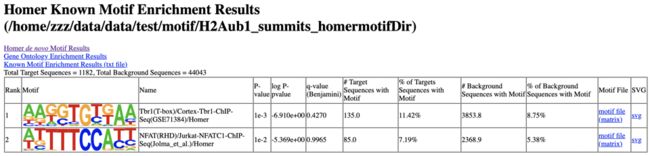
findMotifsGenome.pl ${sample}.bed mm10 ${sample}_homer_motif -len 8,10,12
annotatePeaks.pl ${sample}.bed mm10 1>${sample}.peakAnn.xls 2>${sample}.annLog.txt
done
findMotifs常用参数:
-bg:自定义背景序列;
-size: 用于motif寻找得片段大小,默认200bp;-size given 设置片段大小为目标序列长度;越大需要得计算资源越多;
-len:motif大小设置,默认8,10,12;越大需要得计算资源越多;
-S:结果输出多少motifs, 默认25;
-mis:motif错配碱基数,默认2bp;
-norevopp:不进行反义链搜索motif;
-nomotif:关闭重投预测motif;
-rna: 输出RNA motif,使用RNA motif数据库;
-h:使用超几何检验代替二项式分布;
-N:用于motif寻找得背景序列数目,default=max(50k, 2x input);耗内存参数
参考链接:https://www.jianshu.com/p/93f45acff1f3
不仅仅找了motif,还顺便把peaks注释了一下。得到的后缀为peakAnn.xls 的文件就可以看到和使用R包注释的结果是差不多的(homer official web: http://homer.ucsd.edu/homer/ngs/peakMotifs.html)。
MEME寻找motif
需要通过bed格式的peaks的坐标来获取fasta序列。这个是在线的,我应该不会去用。MEME,链接:http://meme-suite.org/
参考链接:https://www.jianshu.com/p/1384173c353b