自由能计算专题3:gromacs计算自由能的7种方法案例
前言
本教程以甲烷在水中的溶剂化自由能和配体与受体蛋白质结合自由能为入门和进阶例子对使用分子动力学模拟方法来计算自由能的常见方法给出了示例,教程不会过多讲解各种计算自由能方法原理和分析原理,关于自由能计算原理和方法可以参考自由能专题1:原理及常见方法分析原理可以常考自由能专题2:计算与分析指南。该教程全部都经过本人的亲自的模拟和分析,确保无误,从实际操作出发,一步一步手动实现自由能的计算与分析,有助于理解自由能计算原理和结果分析。本教程主要采用GROMACS (5.1.4)分子动力学软件包来进行操作。以下案例的全部模拟配置文件均可在问末的链接中获得。
1.常见自由能计算方法和工具链
分子模拟计算自由能的方法众多,这里简单整理出一个表格,来展示本教程将要运用的自由能计算方法和后处理方法及相关的工具链。如下:
| 计算方法 | 后处理方法 | 自动实现分析工具/脚本 |
|---|---|---|
| PMF | PMF(表达式) | 图表数据处理工具 |
| Umbrella Sampling(US:求解PMF) | WHAM, | gmx wham(gromacs自带) ,PLUMED,Colvars |
| BAR | gmx bar(自带),alchemical_analysis | |
| MBAR(0宽度的WHAM) | alchemical_analysis | |
| FEP(Zwanzig) | DEXP | alchemical_analysis(下同) |
| IEXP | ||
| GINS | ||
| GDEL | ||
| BAR | ||
| UBAR | ||
| RBAR | ||
| MBAR | ||
| TI(Kirkwood) | TI | alchemical_analysis |
| TI-CUBIC | ||
| MM/PBSA | gmx_mm/pbsa(脚本) | |
| g_mm/pbsa(脚本) | ||
| Jarzynski Equality(Jarzynski) | gmx analyze(gromacs自带) | |
| alchemical_analysis | ||
| Metadynamics(MtD) | PLUMED | |
| Colvars | ||
3.自由能计算实例
3.1 PMF:计算甲烷-甲烷之间的平均力势
在本教程中, 我将向您展示如何创建一个包含多个OPLS甲烷的系统, 将其置于TIP4PEW水盒子中, 并根据模拟所得信息计算甲烷-甲烷之间的平均力势.参考Barnett GROMACS教程3: 水中的多个甲烷,既然主题是计算自由能了,那么默认实操者能独立进行简单的分子模模拟任务,并熟知模拟流程,在这里关于模拟所需的三种重要基本配置文件(结构文件(.gro/.pdb), 拓扑文件(.itp,.top)和参数文件(.mdp).)如何创建就不再赘述了。这里会直接给出,只做简单的展示。(下同)
设置
甲烷的拓扑文件如下:
; Topology for methane in TIP4P
#include "oplsaa.ff/forcefield.itp"
#include "oplsaa.ff/tip4pew.itp"
[ moleculetype ]
; Name nrexcl
Methane 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
1 opls_138 1 ALAB CB 1 -0.240 12.011
2 opls_140 1 ALAB HB1 2 0.060 1.008
3 opls_140 1 ALAB HB2 3 0.060 1.008
4 opls_140 1 ALAB HB3 4 0.060 1.008
5 opls_140 1 ALAB HB4 5 0.060 1.008
[ bonds ]
; ai aj funct c0 c1 c2 c3
1 2 1
1 3 1
1 4 1
1 5 1
[ angles ]
; ai aj ak funct c0 c1 c2 c3
2 1 3 1
2 1 4 1
2 1 5 1
3 1 4 1
3 1 5 1
4 1 5 1
; water topology
[ system ]
; Name
Methane in water
[ molecules ]
; Compound #mols
Methane 10
1.首先, 将10个甲烷插入3.2 nm x 3.2 nm x 3.2 nm的盒子中:
$ gmx insert-molecules -ci methane.pdb -o box.gro -nmol 10 -box 3.2 3.2 3.2
2.溶剂化向盒子里加入1000个水分子
$ gmx solvate -cs tip4p -cp box.gro -o conf.gro -p topol.top -maxsol 1000
gmx solvate会自动更新topol.top
3.参数文件
以上过程的所有的文件均可以在1得到:关于PMF配置文件如下:
configura文件:模拟使用的gromacs5.1.4版本
├── methane.pdb :甲烷结构文件
├── box.gro :步骤1生成的文件
├── conf.gro :步骤2生成的文件
├── min1.mdp :第一次能量最小化参数文件
├── min2.mdp :第二次能量最小化
├── eql.mdp :NVT 预平衡
├── eql2.mdp :NPT 预平衡
├── prd.mdp :成品模拟,这里注意你可以改一下时长,我自己仅跑了5ns用于展示,建议跑100ns,rdf更平滑
├── topol.top :拓扑文件,包含10个甲烷和1000个水分子
└── run_pmf.bsh :自动化执行分子模拟流程的脚本
模拟
1.使用run_pmf.bsh脚本进行自动化模拟:
#1.给脚本赋予可执行的权限
$ chmod +x run_pmf.sh
#2.在当前目录下(configura)执行./run_pmf.bash脚本
$ ./run_pmf.sh
分析
1.让我们计算一下所谓的径向分布函数. 首先, 我们需要创建一个索引文件(index.ndx):
#以甲烷中的C原子代表整个分子
$ gmx make_ndx -f conf.gro
> a C
> q
2.计算甲烷-甲烷之间径向分布函数
$ gmx rdf -f prd.xtc -n index.ndx
然后选择 Group 6 “C” 两次,即选择两次6,Ctrl+d 计算生成rdf.xvg 文件
3.安装绘图工具gnuplot,在linux的ubuntu 系统安装:
#在终端输入以下命令自动安装:
$ sudo apt-get install gnuplot
4.绘制RDF(g®)图:
rdf曲线平滑依赖于你保存了多少帧, 运行了多长时间的模拟(原教程运行了100 ns, 保存了25k帧), 如果只是简单地绘制RDF, 得到的结果应该如下所示:(这里放原教程的图)

请注意, 它没有收敛到1. 我们多算了一个, 所以我们应该对其进行修正. 下面是gnuplot命令:
#1.在终端启动gnuplot
$ gnuplot
#2.进行修正:(我们有10个甲烷, 但计算RDF时只用了9个, 所以我们将g的值乘以N/(N-1), 即10/9. 它应该如下所示)
#输入>后面的代码即可
gnuplot> plot 'rdf.xvg' u 1:($2*10/9) w l
修正后的图像:

5.绘制甲烷-甲烷的平均力势(PMF)图:
现在, 要从甲烷-甲烷RDF中得到甲烷-甲烷之间的平均力势, 我们要计算:$ w=−kTln(g)w=−kTln(g).$
因此, 要用gnuplot绘图, 请执行以下操作:
#1.在终端启动gnuplot
$ gnuplot
#2.绘制w(r)
gnuplot> plot 'rdf.xvg' u 1:(-8.314e-3*298.15*log($2*10/9)) w l
得到的图如下:
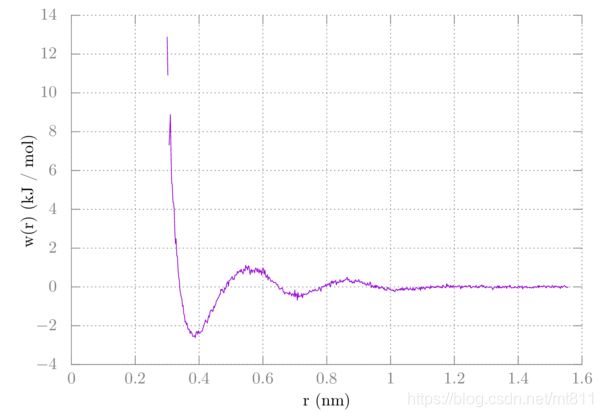
总结
注意到我们的抽样存在一个小的空白区. 这意味着, 在模拟中甲烷没有在那个距离有过相互作用. 我们将在后面的教程中使用伞形采样来解决这个问题。
3.2 Umbrella Sampling(US):使用伞形采样计算甲烷-甲烷的PMF
在本教程中, 我们将使用窗口采样, 有时也称为伞状采样, 来计算甲烷-甲烷的平均力势PMF. 在教程1中, 我们通过模拟几个甲烷分子计算径向分布函数的方法直接获得了PMF. 像这样对几个溶质分子进行采样的方法并不总是可行的.
本教程参考gromacs官方伞形采样(GMX 5.0)和Barnett GROMACS教程5: 使用伞形采样计算甲烷-甲烷的平均力势PMF
模拟所需的甲烷分子拓扑文件如上,同这两个参考教程不同之处是,本教程使用更清晰文件组织来进行模拟,有利于初学者的学习。
设置
1.创建盒子
#将两个甲烷分子插入盒子中
$ gmx insert-molecules -ci methane.pdb -o box.gro -nmol 2 -box 3.2 3.2 3.2
2.溶剂化
$ gmx solvate -cs tip4p -cp box.gro -o conf.gro -p topol.top
3.创建索引文件:
我们还需要创建一个索引文件, 其中包含我们感兴趣的两个组, 它们之间受伞形势限制. 使用gmx make_ndx创建一个索引文件, 其中包含每个甲基碳的组, 并将其分别命名为CA和CB.
$ gmx make_ndx -f conf.gro
> splitres 2 #本人操作是时CH4残基在组2,总之选择CH4残基所在组即可,将残基开成两个CH4(6和7)
> 6 & a C #单独取出两个甲烷分子中的C原子(下同) 生成 8
> 7 & a C #生成 9
> name 8 CA #重命名 8 (下同)
> name 9 CA
>q #保存退出 ,生成index.ndx索引文件
使用gmx make_ndx的其他方法同样可以达到目的. 关键是, 需要将每个甲烷的碳原子放在单独的索引组中, 并分别将它们命名为CA和CB.
4.参数文件
我们在很大程度上重复使用前一个教程(PMF)中的参数文件, 但是我们将添加一个与质心(COM)牵引有关的部分. 牵引代码可以将甲烷维持在指定距离。可能有几种不同的设置方法, 但对于这个系统, 我们将手动指定两个甲烷间的距离.我们将在一般模拟时所需要的所有mdp运行控制文件(能量最小化,npt和nvt预平衡,成品模拟mdp文件)(如1教程 PMF)中添加以下代码:
;{ US采样代码:质心牵引
;===============================================================================
pull = Yes ; Yes/No 使用牵引代码
pull-ngroups = 2 ; 牵引组数目,这里是CA和CB两个
pull-ncoords = 1 ; 只沿一个坐标方向进行牵引
pull-group1-name = CA ; 牵引组名称, 来自top或ndx;.对我们来说,
; 这是其中一个甲烷的碳, 尽管我们也可以选择整个甲烷.
;如果那样做的话, 就会沿着两个甲烷分子的质心方向进行牵引
pull-group2-name = CB ;另一个甲烷的碳原子
pull-coord1-type = umbrella
; umbrella:简谐势
; constraint:刚性约束
; constant-force:恒力, 无须pull-init和pull-rate
pull-coord1-geometry = distance
; distance:沿质心间的矢量, 用pull-dim选择分量
; direction:沿pull-vec方向
; direction-periodic:同direction, 但距离可超过盒长一半. 使用时盒子在牵引方向不能变 化
; cylinder:圆柱
pull-coord1-groups = 1 2 ; 对此坐标牵引的两个组(定义见后面). 实际上你可以定义
;更多的牵引坐标, 也可以在不同的分子组之间进行牵引, 但我们在这里不使用这些选项
pull-coord1-rate = 0.0 ; 速率(nm/ps)
;我们不需要两个组沿坐标方向运动, 因此设置为零
pull-coord1-k = 5000.0 ; 力常数(kJ/mol-nm^2)
pull-coord1-init = WINDOW ; 零时刻的基准距离,这是个变量
;两个组的初始间距. 这是关键字, 我们将使用bash脚本将其替换为每个窗口的相应值
pull-coord1-start = no ; No:不修改pull-init; Yes:将初始构型的质心距离添加到pull-init
;我们手动指定每个窗口的距离, 因此计算时不需要考虑质心距离
;}==============================================================================
5.生成构型文件
为了运行伞形采样, 我们必须沿反应坐标 ζ 生成一系列构型. 其中的一些构型将作为伞形采样窗口的初始构型, 并进行独立的模拟. 下图解释了这些原则. 图的上部显示了我们将要运行的牵引模拟, 目的在于产生一系列沿反应坐标的构型, 模拟完成后会抽取这些构型(图片上部和中部之间的虚线箭头). 图的中部对应着每一采样窗口内的独立模拟, 其中使用伞形偏离势将自由甲烷的质心限制在窗口中. 图的底部展示了构型直方图的理想结果, 相邻窗口存在重叠, 这样以后就可以从这些模拟得到连续的能量函数.

对本教程的例子, 反应坐标为单一方向. 为产生这些构型, 我们必须牵引甲烷CA, 使其远离甲烷CB.我们模拟了26个窗口, 距离从0.05 nm到大约1.3 nm. 请注意, 我为每次模拟都添加了牵引力输出文件选项-pf和牵引距离输出文件选项-px. 这是因为使用-deffnm选项时GROMACS会覆盖这两个文件. 此外, 我使用-n指定了索引文件, 因为gmx grompp需要获取我们使用牵引参数指定的组。
对于其他体系, 保存构型的频率可能需要更高或者更低, 只要足够就好. 关键是沿反应坐标获得足够的构型, 以使伞形采样窗口的间距正常, 窗口间距以甲烷CA和CB的质心(用C来代替了)距离表示, 其中CB为参考组.
由于我们需要为26个窗口进行独立的模拟,那么就需要27份模拟所需要的配置文件,但是我们将使用脚本来简单地更新参数文件模板中的适当值(pull-coord1-init), 因此实际上只需要为模拟过程的每个部分提供一个模板。以上过程所需及生成的文件从1可以获得,其配置文件结构如下:
**configura 文件下的目录结构**
├── collect_dat.sh :批量收集分析所需要数据的脚本
├── batche.sh :批量执行job0.sh -->job26.sh 的脚本
├── write_mdp.sh :批量生成mdp文件的脚本,主要修改mdp 中的pull-coord1-init=WINDOWS 的值
├── write_sh.sh :批量生成job文件的脚本,主要修改job文件中 WINDOW=Wnum的值,
├── job.sh :为每一窗口独立运行整个模拟过程(EM->NVT->NPT->PRD),即WINDOW0,
├── MDP :总运行参数mdp文件夹
│ ├── EM :能量最小化参数mdp文件夹
│ │ ├── 1em.mdp :第1次能量最小化参数mdp模板文件
│ │ ├── 2em.mdp :第2次能量最小化参数mdp模板文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh 1em 即可后接mdp文件前缀 ./write_mdp.sh 2em
│ ├── NVT :等温等容预平衡参数mdp文件夹
│ │ ├── nvt.mdp :等温等容预平衡参数mdp模板文件
│ │ ├── write_mdp.sh : 用法:./write_mdp.sh nvt 即可后接mdp文件前缀
│ ├── NPT :等温等压预平衡参数mdp文件夹
│ │ ├── npt.mdp :等温等压预平衡参数mdp模板文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh npt 即可后接mdp文件前缀
│ └── PRD :成品模拟参数mdp文件夹
│ ├── prd.mdp :成品模拟参数模板mdp文件
│ └── write_mdp.sh : 用法:./write_mdp.sh prd 即可后接mdp文件前缀
├── Methane :结构文件和拓扑文件夹
│ ├── box.gro :10个甲烷盒子结构文件
│ ├── conf.gro :模拟体系结构文件10个甲烷+水
│ ├── index.ndx :甲烷中所有原子的索引文件
│ ├── methane.pdb :甲烷结构文件
│ └── topol.top :体系top文件
└── REDERME.md
5.1.使用write_mdp.sh脚本在文件夹EM/NVT/NPT/PRD下分别生成0到26个.mdp文件
脚本如下:
#!/bin/bash
#对于模拟, 我们将使用shell脚本替换.mdp文件中的WINDOW关键字
# 打开一个通用的.mdp文件,并替换以下值
#26个窗口, 距离从0.05 nm到大约1.3 nm.既WINDOW有27个值,增量为0.05
for ((i=0;i<27;i++))
do
# 小数点前补0
x=$(echo "0.05*$(($i+1))" | bc |awk '{printf "%.2f",$0}');
sed 's/WINDOW/'$x'/g' $1.mdp >$1$i.mdp
done
使用示例:例如在./configura/MDP/EM下批量生成1em.mdp和2em.mdp各27份
#给脚本赋予可执行权限
$chmod +x write_mdp.sh
#批量生成27份1em.mdp文件
$ ./write_mdp.sh 1em
#批量生成27份2em.mdp文件
$ ./write_mdp.sh 2em
接下来以上述方式生成整个模拟所需要的mdp文件(同上)
模拟
1.对于每一个独立窗口我们可以用脚本(job.sh)进行自动化的模拟整个过程如下:
#!/bin/bash
# 设置一些环境变量,即你所有模拟配置文件所在的根目录(里面包括MDP,Methane,.sh脚本,在命令行使用pwd即可获得)
#"/home/eric/free_energy/04.US/configura" 即为我所有模拟配置文件所在根目录
FREE_ENERGY=/home/eric/free_energy/04.US/configura
echo "Free energy home directory set to $FREE_ENERGY"
MDP=$FREE_ENERGY/MDP
echo ".mdp files are stored in $MDP"
#Wnum 为窗口数,将会被write_sh.sh进行批量替换成27个不同的值
WINDOW=Wnum
# 将为WINDOW的每个值和工作流程中的每个步骤创建一个新的目录,以实现最大限度的组织。
mkdir Window_$WINDOW
cd Window_$WINDOW
#################################
# 能量最小化第1步:STEEP #
#################################
echo "Starting minimization for Window = $WINDOW..."
mkdir EM_1
cd EM_1
# Iterative calls to grompp and mdrun to run the simulations
gmx grompp -f $MDP/EM/1em$WINDOW.mdp -c $FREE_ENERGY/Methane/conf.gro -p $FREE_ENERGY/Methane/topol.top -n $FREE_ENERGY/Methane/index.ndx -o min$WINDOW.tpr -maxwarn 1
gmx mdrun -nt 2 -v -deffnm min$WINDOW -pf pullf-min$WINDOW -px pullx-min$WINDOW
sleep 10
#################################
# 能量最小化第 2步: STEEP #
#################################
cd ../
mkdir EM_2
cd EM_2
#我们在这里使用了 -maxwarn 1,因为 grompp 错误地抱怨使用了一个普通的截止值。
# 这是一个小问题,将在未来的Gromacs版本中得到修正。
gmx grompp -f $MDP/EM/2em$WINDOW.mdp -c ../EM_1/min$WINDOW.gro -p $FREE_ENERGY/Methane/topol.top -n $FREE_ENERGY/Methane/index.ndx -o min$WINDOW.tpr -maxwarn 1
gmx mdrun -nt 2 -v -deffnm min$WINDOW -pf pullf-min$WINDOW -px pullx-min$WINDOW
echo "Minimization complete."
sleep 10
#####################
# NVT 等容平衡 #
#####################
echo "Starting constant volume equilibration..."
cd ../
mkdir NVT
cd NVT
gmx grompp -f $MDP/NVT/nvt$WINDOW.mdp -c ../EM_2/min$WINDOW.gro -p $FREE_ENERGY/Methane/topol.top -n $FREE_ENERGY/Methane/index.ndx -o nvt$WINDOW.tpr -maxwarn 1
gmx mdrun -nt 2 -v -deffnm nvt$WINDOW -pf pullf-nvt$WINDOW -px pullx-nvt$WINDOW
echo "Constant volume equilibration complete."
sleep 10
#####################
# NPT 等压平衡 #
#####################
echo "Starting constant pressure equilibration..."
cd ../
mkdir NPT
cd NPT
gmx grompp -f $MDP/NPT/npt$WINDOW.mdp -c ../NVT/nvt$WINDOW.gro -p $FREE_ENERGY/Methane/topol.top -n $FREE_ENERGY/Methane/index.ndx -t ../NVT/nvt$WINDOW.cpt -o npt$WINDOW.tpr -maxwarn 1
gmx mdrun -nt 2 -v -deffnm npt$WINDOW -pf pullf-npt$WINDOW -px pullx-npt$WINDOW
echo "Constant pressure equilibration complete."
sleep 10
#################
# 成品模拟 MD #
#################
echo "Starting production MD simulation..."
cd ../
mkdir Production_MD
cd Production_MD
gmx grompp -f $MDP/PRD/prd$WINDOW.mdp -c ../NPT/npt$WINDOW.gro -p $FREE_ENERGY/Methane/topol.top -n $FREE_ENERGY/Methane/index.ndx -t ../NPT/npt$WINDOW.cpt -o prd$WINDOW.tpr -maxwarn 1
gmx mdrun -nt 2 -v -deffnm prd$WINDOW -pf pullf-prd$WINDOW -px pullx-prd$WINDOW
echo "Production MD complete."
# End
echo "Ending. Job completed for Window= $WINDOW"
2.对于以上脚本你仅需要修改:FREE_ENERGY=/home/eric/free_energy/04.US/configura 中的“/home/eric/free_energy/04.US”部分。这一部分为你配置文件configura所在根目录,每个人都是不一样的。
3.以上只是一个独立窗口的完整的模拟脚本,而我们需要模拟27次,即需要下列脚本(write_sh.sh )批量生成27个job.sh脚本:
#!/bin/bash
#对于模拟, 我们将使用shell脚本替换.job文件中的Wnum关键字
# 打开一个通用的job.sh文件,并替换Wnum的值
#26个窗口, 即Wnum有26个值,从0到26
for ((i=0;i<27;i++))
do
sed 's/Wnum/'$i'/g' $1.sh >$1$i.sh
done
- 准备工作全部做完后如下开始进行27个窗口独立的模拟,使用一个简单脚本( batche.sh)批量自动化这一过程如下:
#!/bin/bash
#批量执行job脚本,这里0时已经被测式执行来。从1开始
#给新生成的26个Job脚本赋予可执行的权限
chmod +x job*.sh
for num in {0..26}
do
./job$num.sh
done
5.开始模拟
#1.赋予脚本执行权限
$ chmod +x batche.sh
#2.开始全部的模拟(共27,这里只是用单机电脑来跑的,如果使用集群可以27个任务并行运行,出结果更快)本机4核8线程差不多三天完成。
$ ./batche.sh
分析
1.正常模拟结束将会产生27个Window_0 到Window_27文件夹,该文件夹内容如下,以Window_0为例:
├── Window_0
│ ├── EM_1
│ │ ├── min0.gro
│ │ └── ....
│ ├── EM_2
│ │ ├── min0.gro
│ │ └── ....
│ ├── NVT
│ │ ├──nvt0.gro
│ │ └── .....
│ ├── NPT
│ │ ├── npt0.gro
│ │ └── .....
│ └── PRD
│ ├── prd0.gro
│ ├── prd.tpr ;分析所需文件
│ ├── pullf-prd0.xvg ;分析所需文件
│ └── ....
2.收集26个窗口,即27份独立的模拟成品模拟文件,使用一个简单脚本(collect_dat.sh)将27Window文件夹下的数据收集起来如下:
#!/bin/bash
#创建保存分析需要用的prd0.tpr 到prd26.tpr 和 pullf-prd0.xvg 到pullf-prd26.xvg文件的文件夹 analysis_us
mkdir analysis_us
cd analysis_us
for num in {0..26}
do
cp ../Window_$num/Production_MD/prd$num.tpr ./
cp ../Window_$num/Production_MD/pullf-prd$num.xvg ./
done
3.我们将使用gmx wham来获取PMF. 该程序接受一个包含.tpr文件列表的文件和另一个包含.xvg文件列表的文件, 后者中包含了输出的牵引力.要创建这两个文件列表的文件, 在analysis_us文件夹下执行以下命令:
$ ls prd.*.tpr > tpr.dat
$ ls pullf-prd.*.xvg > pullf.dat
4.使用GROMACS实现的加权直方图分析(WHAM)来重建PMF:
$ gmx wham -it tpr.dat -f pullf.dat
运行gmx wham之后, 平均力势的计算结果保存在名为profile.xvg的文件中. 绘图应该如下所示:
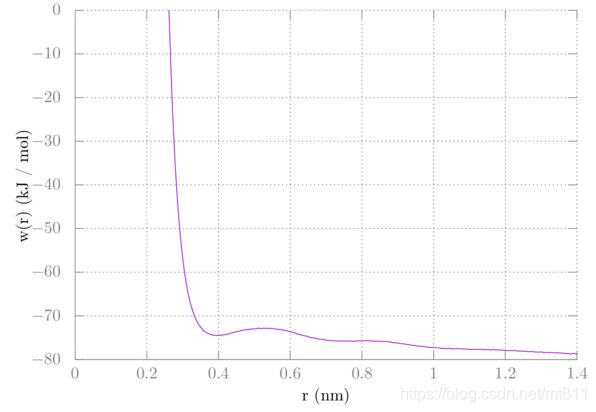
5.图像修正
我们希望在较远的距离处相互作用为零. 但是, 由于我们使用了三维的偏置势, 需要进行校正. 想象一下将其中一个甲烷作为参考点. 另一个甲烷可以在该点周围距离 r处进行采样, 覆盖了半径为 rr的某个球体的表面. 这为我们的采样增加了额外的构型空间, 降低了熵. 需要消除这些额外熵对PMF的贡献. 回想一下, 等温等压系综中的吉布斯自由能是 −kTln(W), 其中 W 为分配函数. 对我们的情况, 甲烷绕着球体表面采样时, w 与该球体的表面积成正比. 因此, 需要增加 2kTln® 的校正. 此外, 我们需要将图形向上移动, 使其尾部变为零. 对我的结果需要增加的数值大约为7, 但你需要增加的数值可能不同. 要在gnuplot中绘制此图, 请在gnuplot终端中执行以下操作:
#1.在终端启动gnuplot
$ gnuplot
#2.进行修正:输入>后面的代码即可
gnuplot> plot 'profile.xvg' u 1:($2+2*8.314e-3*298.15*log($1)+77) w l
您的PMF现在应该如下所示:

总结
与教程1中的PMF相比, 可以看到它们几乎相同:

区别在于, 使用教程3的直接方法, 采样不可能接近窗口采样. 两个甲烷不会自然地相互靠近, 这就是为什么我们必须增加伞形势以维持它们之间距离的原因。
另一个输出是histo.xvg, 它有助于确定窗口之间是否有足够的重叠. 以下是我模拟的每个直方图的绘图:

很显然, 我们的窗口已经足够重叠了. 否则的话, 我们可能必须选择较小的窗口, 或选择缺少模拟的特定点进行模拟.
3.3 FEP:计算甲烷在水中的溶剂化自由能
该教程在网上三个类似的教程,笔者在这里先简单例举出来,然后再说明这些教程的异同,和参考这些教程哪些内容
| 参考教程链接 参考内容 | ||
|---|---|---|
| 教程1:gromacs 官方教程6: 自由能计算流程,分析方法(bar) | ||
| 教程2:译:自由能计算: 水中的甲烷(GMX 5.0) 与教程1其他都相同,算升级版,多分析了两组数据 | ||
| 教程3:译:Barnett GROMACS教程4: 甲烷的溶剂化自由能 使用python脚本对自由能计算方法自动化分析 | ||
教程3和教程(1和2)不同之处有四点:
一是前者使用水模型同3.1和3.2一样是TIP4P,而后者是TIP3P;
二是前者在描述自由能的变化时考虑了范德华和静电相互作用,而后者只考虑范德华相互作用;
三是前者使用分析自由能的方法不同,前者是TI,后者是使用了gromacs自带的BAR后处理方法;
四是前者只进行了15次独立模拟,后者进行21次独立模拟。
根据FEP和TI计算原理可知,使用分子模拟计算自由能时都是通过多个 λ \lambda λ值控制不同相互作用类型的进度来处理自由能计算,因此这样就可以分别控制库仑、LJ和约束变换等自由能计算,所以无论使用FEP和TI那一种计算方法,在gromacs它们使用的都是同一套自由能计算代码,只是对自由能的后处理分析不同而已。
本教程主要基于教程(1和2)模拟设置进行模拟,,将指导用户计算一个简单的自由能变化, 中性甲烷与一盒子水之间范德华相互作用的去耦合(即移除),并使用gromacs自带工具和python分析工具(alchemical_analysis)中基于FEP计算自由能的8种后处理方法来简要分析这一过程中自由能的变化,会涉及相关工具的使用和安装。
体系从状态A转变到状态B过程的自由能变化 Δ G A B \Delta G_{AB} ΔGAB是耦合参数 λ \lambda λ的函数, 它代表了状态A和B之间转变的程度, 也就是哈密顿量的微扰程度以及体系已经变化的程度. 利用不同 λ \lambda λ 对应的模拟可以绘制 ∂ H / ∂ λ \partial H/\partial \lambda ∂H/∂λ曲线, 并由此得到 Δ G A B \Delta G_{AB} ΔGAB自由能计算的第一步是确定使用多少个点描述从状态A( λ \lambda λ= 0)到状态B( λ = 1 \lambda = 1 λ=1)的转变, 其目的在于收集足够多的数据, 完全充分地对相空间进行取样, 并得到合理的 ∂ H / ∂ λ \partial H/\partial \lambda ∂H/∂λ 曲线:

如上图所示,状态A是在水中完全相互作用的甲烷. 状态B将是一个甲烷被隐去, 表示它与水的所有范德华相互作用和库仑相互作用(本例只考虑范德华力)都被关闭; 甲烷可以与自身相互作用, 水可以与自身相互作用, 但甲烷和水不会相互作用. 就好像我们将甲烷从水中取出并置于真空中, 而在其他地方我们仍然拥有甲烷所在的水. 我们总共有21个状态。
设置
1.准备甲烷的拓扑文件:注意此模拟使用的TIP3P水模型
; Topology for methane in TIP3P
#include "oplsaa.ff/forcefield.itp"
[ moleculetype ]
; Name nrexcl
Methane 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
1 opls_138 1 ALAB CB 1 0.000 12.011
2 opls_140 1 ALAB HB1 2 0.000 1.008
3 opls_140 1 ALAB HB2 3 0.000 1.008
4 opls_140 1 ALAB HB3 4 0.000 1.008
5 opls_140 1 ALAB HB4 5 0.000 1.008
[ bonds ]
; ai aj funct c0 c1 c2 c3
1 2 1
1 3 1
1 4 1
1 5 1
[ angles ]
; ai aj ak funct c0 c1 c2 c3
2 1 3 1
2 1 4 1
2 1 5 1
3 1 4 1
3 1 5 1
4 1 5 1
; water topology
#include "oplsaa.ff/tip3p.itp"
[ system ]
; Name
Methane in water
[ molecules ]
; Compound #mols
Methane 1
SOL 596
你会注意到, 所有的电荷都设置为零. 这种设置有一个实际的原因, 正常情况下, 在关闭范德华相互作用之前, 会先关闭溶质与水之间的电荷相互作用. 如果仅仅保留电荷相互作用而关闭了Lennard-Jones项, 正负电荷相互接近的距离可能无限小, 导致体系非常不稳定, 并可能崩溃. 本教程的步骤基本假定在此之前已经正确地关闭了电荷项, 我们将只关闭溶质与溶剂分子之间的范德华相互作用.这一点下面在设置自由能计算参数(couple-lambda0 )会提到。
2.参数文件:
同教程3.2.US类似,只是我们添加的是一个计算自由能部分而前者是伞形拉伸的部分, 以便慢慢关闭甲烷与水的相互作用。
另外, 我们还需要为每个 λ \lambda λ状态的准备独立模拟所必备的基本配置文件. 我们需要21个最小化, 21个预平衡,21个成品模拟等的参数文件. 但是我们将使用脚本来简单地更新参数文件模板中的适当值(init_lambda_state ), 因此实际上只需要为模拟过程的每个部分提供一个模板。
以em_steep.mdp模板为例:(前部分为一般模拟所学控制参数,请重点关注自由能计算部分控制参数!!!)
; ;{ 预处理, 使用 C++ 语法
;===============================================================================
include = ; 含引用文件的目录, 拓扑文件中可引用其中的文件, 可多项
; 例: -I/home/joe/joy -I/home/tom/tim -IC:/GMX/top
define = -DFLEXIBLE
; 预定义, 默认无, 可多项, 区分大小写
; -DPOSRES: 使用位置限制文件进行位置限制动力学模拟
; -DFLEXIBLE: 启用柔性水, steep效果更好, cg, l-bfgs或简正分析须开启
;}==============================================================================
;{ 能量最小化运行控制
;===============================================================================
integrator = l-bfgs ;能量最小化方法, steep:最陡下降; cg:共轭梯度; lbfgs:二次收敛
nsteps = 5000 ; 最大积分步数, 默认0, -1:无限制
emtol = 100 ; 力的最大容差(kJ/mol-nm), 对壳层值应小于1
emstep = 0.01 ; 初始步长(nm)
nbfgscorr = 10 ; L-BFGS最小化的校正步数
;}==============================================================================
;{ 壳层动力学
;===============================================================================
niter = 20 ; 优化壳层位置和柔性约束的最大迭代次数
;}==============================================================================
;{ 输出控制
;===============================================================================
nstlog = 1 ; 日志文件输出频率
nstenergy = 1 ; 能量文件输出频率
;}==============================================================================
;{ 邻区搜索
;===============================================================================
cutoff-scheme = verlet ; 截断方式, Verlet:粒子截断; Group:电荷组
nstlist = 1 ; 邻区列表更新频率, 0:真空模拟; -1:自动
ns_type = grid ; 邻区搜索算法, Grid:较快; Simple:仅与Group联用
rlist = 1.2 ; 邻区列表截断距离(nm)
rcoulomb = 1.2 ; 静电截断半径, 不超过最小盒子边长一半
rvdw = 1.2 ; 范德华截断半径
;}==============================================================================
;{ 周期性
;===============================================================================
pbc = XYZ ; 周期性边界条件, XYZ; XY; No:忽略盒子, 截断与nstlist置零
;}==============================================================================
;{ 静电与范德华
;===============================================================================
; Electrostatics
coulombtype = PME ; 静电计算方法
; van der Waals
vdwtype = cutoff ; 范德华计算方法
vdw-modifier = potential-switch ; ;修正方法
rvdw-switch = 1.0 ;从何时开始切换LJ力或势能,只适用与对力或势能切换的情况
DispCorr = EnerPres ; 长程色散校正, No:无; Ener:能量; EnerPres:能量和压力
;}==============================================================================
;{ EWALD/PME/PPPM
;===============================================================================
fourierspacing = 0.12 ;PME/PPPM方法FFT格点最大间距(nm)
pme_order = 6 ;内插阶数, 默认4相当于三次插值
ewald_rtol = 1e-06 ; 静电能量容差
epsilon_surface = 0 ; 偶极校正
;}==============================================================================
;{ 温度和压力耦合,在能量最小化时被关闭,可以不写此参数
;===============================================================================
tcoupl = no
pcoupl = no
;}==============================================================================
;{ 初始速度 ,能量最下化时没有速度,该参数可不写
;===============================================================================
gen_vel = no
;}==============================================================================
;{ 键约束
;===============================================================================
constraints = none ; None:仅拓扑中指定的约束 L-BFGS 在此不能使用约束
; h-bonds:含氢键; h-angles:含氢键和键角
; all-bonds:所有键; all-angles:所有键和键角
constraint-algorithm = lincs ; 约束算法, Lincs:不支持键角; Shake:慢且不稳定, 不支持EM和dd
continuation = no ; 是否约束初始构型, 并重置壳层
lincs-order = 12 ; 约束耦合矩阵展开的最高阶数
;{ 自由能计算
;===============================================================================
free_energy = yes ; No/Yes/Expand,表示进行自由能计算, 将会进行所选分子(下面定义)从状态A到状态B的内插
init_lambda_state = nlambda ;向量的一个索引号(始于0),它只是一个整数的占位符,我们将模拟21个不同 ;的状态, 因此这个数字的范围是0到20.
;我们的脚本会将其替换为每个状态相应的值(0,,,20)
delta_lambda = 0 ; 每步的增量, 慢增长方法取非零值
calc_lambda_neighbors = -1 ;相邻的数目. bar取1, mbar取-1,教程1和2都是取1,这里我们需要使用mbar
;就取-1了,方便我们使用所有分析方法,一站式处理。
;此处指定的lambda矢量,每一个组合都是一个索引,从init_lambda_state中为每一个模拟检索。
; init_lambda_state 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
vdw_lambdas = 0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.40 0.45 0.50 0.55 0.60 0.65 0.70 0.75 0.80 0.85 0.90 0.95 1.00 ;用于范德华相互作用转变的 λ 值的数组
coul_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 ;用于库伦(静电)相互作用转变的 λ 值的数组
;我们不会改变任何键合或约束的相互作用。
bonded_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 ;用于键合相互作用转变的 λ 值的数组
restraint_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 ;用于限制相互作用转变的 λ 值的数组
;质量不变(lambda=0和lambda=1时粒子特性相同)。
mass_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 ;用于原子质量转变的 λ 值的数组; 当转变分子的化学本质时会使用.
;温度不变,这里不做模拟退火
temperature_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 ;用于温度转变的 λ 值的数组; 仅用于模拟退火
; 解耦选项
sc-alpha = 0.5 ;“软核”Lennard-Jones方程中使用的 α 缩放因子的值
sc-coul = no ; 线性地转变库伦相互作用. 这是默认行为, 给出是为了更清楚
sc-power = 1 ;软核方程中 λ 的次数
sc-sigma = 0.3 ;对C6或C12参数为0, 或者 σ < sc-sigma(典型为H原子)的任意原子类型, 赋给 σ 的值
couple-moltype = Methane ; 耦合的分子类型, 使用拓扑中的名称
couple-lambda0 = vdw ;当lambda为0(状态A)时, VDW完全打开
; 作用类型, None:无; vdw-q:所有; vdw:仅静电 :A状态有。
;如果要使用vdw-q,话那么我们必须给甲烷分子指定电荷,可能导致不稳定
;表示该过程为去耦合,从有到无一系列的变化
couple-lambda1 = none ;当lambda为1(状态B)时, 作用类型, None:无; vdw-q:所有; vdw:仅vdw;
couple-intramol = no ; 不对分子内相互作用进行去耦合. 也就是说, λ 因子只用于溶质溶剂之间的非键相互作用, 而不用于溶质溶质之间的非键相互作用.
nstdhdl = 100 ;H/λ 和 ΔH 输出到dhdl.xvg文件的频率.
separate-dhdl-file =yes ;输出单独的dhdl.xvg文件
;===============================================================================
3.模拟所需要的基本配置文件已经全部备好,无需再让你新建立盒子溶剂化,这两步直接省略,文件目录同教程3.2基本差不多如下:
**configura 文件下的目录结构**
├── collect_dhdl.sh :批量收集分析所需要数据的脚本
├── batche.sh :批量执行job_0.sh -->job_20.sh 的脚本
├── write_mdp.sh :批量生成mdp文件的脚本,主要修改mdp 中的init_lambda_state =nlambda 的值
├── write_sh.sh :批量生成job文件的脚本,主要修改job文件中 LAMBDA=Wnum的值,
├── job.sh :为每一窗口独立运行整个模拟过程(EM->NVT->NPT->Production_MD),即Lambda_0,
├── MDP :总运行参数mdp文件夹
│ ├── EM :能量最小化参数mdp文件夹
│ │ ├── em_l-bfgs.mdp :第1次能量最小化参数mdp文件
│ │ ├── em_steep.mdp :第2次能量最小化参数mdp文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh em_l-bfgs
│ ├── NVT :等温等容预平衡参数mdp文件夹
│ │ ├── nvt.mdp :等温等容预平衡参数mdp文件
│ │ ├── write_mdp.sh : 用法:./write_mdp.sh nvt 即可后接mdp文件前缀
│ ├── NPT :等温等压预平衡参数mdp文件夹
│ │ ├── npt.mdp :等温等压预平衡参数mdp文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh npt 即可后接mdp文件前缀
│ └── Production_MD :成品模拟参数mdp文件夹
│ ├── md.mdp :成品模拟参数mdp文件
│ └── write_mdp.sh : 用法:./write_mdp.sh md 即可后接mdp文件前缀
├── Methane :结构文件和拓扑文件夹
│ ├── methane.gro :含1个甲烷水盒子结构文件
│ └── topol.top :体系top文件
└── REDERME.md
4.为每一个独立 λ \lambda λ状态生成模拟所学基本配置文件,操作过程和上诉3.2 US教程类似,这里不在赘述,只是使用脚本替换的关键词不同而已,如下:
#!/bin/bash
#对于模拟, 我们将使用shell脚本替换.mdp文件中的nlambda关键字
# 打开一个通用的.mdp文件,并替换nlambda的值
#合计21个状态值
for ((i=0;i<21;i++))
do
sed 's/nlambda/'$i'/g' $1.mdp >$1_$i.mdp
done
模拟
在每个 λ \lambda λ状态下, 我们将进行两次能量最小化(bfgs/steep), NVT预平衡100 ps, NPT预平衡100ps, 然后进行1ns的成品模拟.
需要生成21个模拟工作流自动化执行脚本文件(job_0.sh ,job_20.sh),然后进行批量模拟,过程同上述教程3.2 US模拟操作过程一样,这里不再赘述了。
分析
1.正常模拟结束将会产生21个Lambda_0 到Lambda_20文件夹,该文件夹内容如下,以Lambda_0为例:
├── Lambda_0
│ ├── EM_1
│ │ ├── min0.gro
│ │ └── ....
│ ├── EM_2
│ │ ├── min0.gro
│ │ └── ....
│ ├── NVT
│ │ ├──nvt0.gro
│ │ └── .....
│ ├── NPT
│ │ ├── npt0.gro
│ │ └── .....
│ └── PRD
│ ├── md0.gro
│ ├── md0.tpr
│ ├── dhdl.xvg ;分析所需文件,在成品模拟时,
│ └── ....
2.关于生成分析所需要的数据文件(如:dhdl.xvg文件),需要在参数文件成品模拟mdp,和运行模拟时设置如下(以本例为例):
#1.独立生dhdl文件,需要注意以下两步:(本教程的做法)
(1)在md.mdp中设置以下两项:
nstdhdl = 100 ; ;∂H/∂λ 和 ΔH 输出到dhdl.xvg文件的频率
separate-dhdl-file =yes ;输出单独的dhdl.xvg文件
(2)在运行成品模拟是设置 -dhdl选项,如下:(在脚本job_0中)
gmx mdrun -nt 2 -v -deffnm md0 -dhdl dhdl
#2.默认输出,从md0.edr文件中获取dhdl.xvg文件: (替代做法)
(1)在md.mdp中设置以下两项:
nstdhdl = 100 ;∂H/∂λ 和 ΔH 输出到dhdl.xvg文件的频率
separate-dhdl-file =no ;自由能的值输出到能量输出文件(ener.edr,以累积块的形式,每 nstenergy步一次)
(2)在运行成品模拟是设置如下:
gmx mdrun -nt 2 -v -deffnm md0
(3)gmx energy 从md0.edr文件中获取dhdl.xvg
gmx energy -f md0.edr -s md0.tpr -odh -dhdl.xvg
#3.gromacs官网的例子:<自由能的计算:水中甲烷> 设置好如下 (教程1和2做法)
(1)在md.mdp中设置仅一项:
nstdhdl = 100 ;∂H/∂λ 和 ΔH 输出到dhdl.xvg文件的频率
(2)在运行成品模拟是设置如下:
gmx mdrun -nt 2 -v -deffnm md0
#(没设置 separate-dhdl-file=no ∂H/∂λ 和 ΔH 值不会输出到.edr文件中),仅生成md0.xvg文件
3.官网的教程教程1和翻译的教程2是默认每一个$ \lambda 不 独 立 生 成 d h d l . x v g 文 件 ( 含 不独立生成dhdl.xvg文件(含 不独立生成dhdl.xvg文件(含\langle \frac{ \partial H} { \partial \lambda} \rangle 和 和 和\Delta H 值 ) , 而 是 生 成 了 一 个 m d 0. x v g 该 文 件 其 实 是 包 括 d h d l . x v g 文 件 的 数 据 , 可 以 使 用 g m x b a r , 快 捷 的 分 析 值),而是生成了一个md0.xvg 该文件其实是包括dhdl.xvg文件的数据,可以使用gmx bar,快捷的分析 值),而是生成了一个md0.xvg该文件其实是包括dhdl.xvg文件的数据,可以使用gmxbar,快捷的分析\Delta G_{AB}$
4.为了更加直观展示基于FEP计算自由能的不同分析方法,我尝试2.中的三种做法,模拟过程都一样,只不过改了些相关参数,生成了不同的数据文件,这里不再赘述。这里只重点讲分析
| 分析工具 | 分析输入数据文件 | |
|---|---|---|
| dhdl*.xvg (2中1,2) | md*.xvg(2中3) | |
| gmx bar | 1 | 2 |
| alchemical_analysis | 3 | 4 |
本例子使用的参数文件是采用2中1的方式生成的 21个 λ \lambda λ状态的dhdl.xvg系列文件(dhdl.xvg)作为分析输入文件, ⟨ ∂ H ∂ λ ⟩ \langle \frac{ \partial H} { \partial \lambda} \rangle ⟨∂λ∂H⟩(用来基于TI来进行分析)和 Δ H \Delta H ΔH(用来基于FEP来进行分析)值。两者H代表哈密顿值,对于孤立体系来说哈密顿量等于能量,及又可以表示为 ⟨ ∂ U ∂ λ ⟩ \langle \frac{ \partial U} { \partial \lambda} \rangle ⟨∂λ∂U⟩和 Δ U \Delta U ΔU,2中的其他两种做法,这里仅分析,如使用md.xvg系列文件(md.xvg)。
如上表所示 根据分析工具不同和分析输入文件不同,可以组合成四种不同的分析模式,实则两两相同,下面一一道来。
使用gmx bar 工具分析:
1.使用gmx bar 工具分析dhdl.xvg系列数据数据:(本教程分析方法)
#使用gmx bar 读入单独生成的dhdl.xvg系列文件或者是从md.edr系列文件提取出的dhdl.xvg:dhdl0.zvg---dhdl21.xvg
gmx bar -f dhdl*.xvg -o -oi -oh
屏幕打印出下列信息:
emperature: 300 K
Detailed results in kT (see help for explanation):
#A点的λ值 B点的λ值 自由能估计 B在A中的相对熵估计 A在B中的相对熵估计 每个样本标准偏差的估计期望
lam_A lam_B DG +/- s_A +/- s_B +/- stdev +/-
0 1 0.07 0.00 0.03 0.00 0.04 0.00 0.25 0.00
1 2 0.06 0.00 0.03 0.00 0.03 0.00 0.26 0.00
2 3 0.05 0.00 0.03 0.00 0.04 0.00 0.27 0.00
3 4 0.03 0.00 0.03 0.00 0.04 0.00 0.27 0.00
4 5 0.02 0.00 0.04 0.00 0.05 0.00 0.29 0.00
5 6 -0.00 0.01 0.04 0.00 0.05 0.00 0.30 0.01
6 7 -0.03 0.00 0.06 0.01 0.07 0.01 0.33 0.00
7 8 -0.07 0.00 0.05 0.01 0.07 0.01 0.36 0.00
8 9 -0.10 0.00 0.06 0.00 0.08 0.00 0.37 0.00
9 10 -0.16 0.01 0.09 0.01 0.12 0.01 0.42 0.00
10 11 -0.24 0.01 0.09 0.01 0.12 0.01 0.46 0.01
11 12 -0.32 0.01 0.11 0.01 0.15 0.01 0.51 0.01
12 13 -0.43 0.01 0.15 0.01 0.18 0.01 0.57 0.01
13 14 -0.56 0.01 0.16 0.01 0.17 0.01 0.56 0.01
14 15 -0.58 0.01 0.09 0.01 0.09 0.01 0.46 0.00
15 16 -0.51 0.01 0.07 0.01 0.07 0.01 0.35 0.00
16 17 -0.40 0.00 0.03 0.00 0.03 0.00 0.26 0.00
17 18 -0.28 0.00 0.02 0.00 0.02 0.00 0.19 0.00
18 19 -0.16 0.00 0.01 0.00 0.01 0.00 0.13 0.00
19 20 -0.05 0.00 0.00 0.00 0.00 0.00 0.10 0.00
#注意 1Kcal/mol=4.184KJ/mol; KT(T=300K)=0.6kcal/mol
Final results in kJ/mol:
point 0 - 1, DG 0.17 +/- 0.01
point 1 - 2, DG 0.14 +/- 0.01
point 2 - 3, DG 0.12 +/- 0.01
point 3 - 4, DG 0.09 +/- 0.01
point 4 - 5, DG 0.05 +/- 0.01
point 5 - 6, DG -0.00 +/- 0.01
point 6 - 7, DG -0.08 +/- 0.01
point 7 - 8, DG -0.16 +/- 0.01
point 8 - 9, DG -0.24 +/- 0.01
point 9 - 10, DG -0.40 +/- 0.02
point 10 - 11, DG -0.59 +/- 0.02
point 11 - 12, DG -0.79 +/- 0.02
point 12 - 13, DG -1.08 +/- 0.03
point 13 - 14, DG -1.39 +/- 0.03
point 14 - 15, DG -1.45 +/- 0.03
point 15 - 16, DG -1.28 +/- 0.01
point 16 - 17, DG -1.01 +/- 0.01
point 17 - 18, DG -0.70 +/- 0.01
point 18 - 19, DG -0.39 +/- 0.01
point 19 - 20, DG -0.12 +/- 0.00
total 0 - 20, DG -9.11 +/- 0.06
2.使用gmx bar 工具分析md.xvg系列数据数据:(教程3分析方法)
#使用gmx bar 读入生成的md0.xvg系列文件md0.xvg---md21.xvg
gmx bar -f md*.xvg -o -oi -oh
屏幕打印出下列信息:
Temperature: 300 K
Detailed results in kT (see help for explanation):
lam_A lam_B DG +/- s_A +/- s_B +/- stdev +/-
0 1 0.07 0.00 0.03 0.00 0.03 0.00 0.25 0.00
1 2 0.06 0.00 0.03 0.00 0.04 0.00 0.26 0.00
2 3 0.05 0.00 0.03 0.00 0.04 0.00 0.27 0.00
3 4 0.03 0.00 0.03 0.00 0.04 0.00 0.28 0.00
4 5 0.01 0.00 0.04 0.00 0.05 0.00 0.30 0.00
5 6 -0.01 0.00 0.04 0.00 0.05 0.00 0.31 0.00
6 7 -0.03 0.00 0.05 0.00 0.06 0.00 0.32 0.00
7 8 -0.06 0.00 0.06 0.00 0.07 0.00 0.35 0.00
8 9 -0.10 0.00 0.06 0.00 0.08 0.00 0.38 0.01
9 10 -0.15 0.00 0.07 0.00 0.10 0.00 0.42 0.00
10 11 -0.23 0.01 0.10 0.00 0.13 0.01 0.46 0.01
11 12 -0.33 0.01 0.12 0.01 0.16 0.01 0.52 0.01
12 13 -0.46 0.01 0.16 0.01 0.19 0.01 0.58 0.00
13 14 -0.57 0.01 0.14 0.01 0.16 0.01 0.56 0.00
14 15 -0.59 0.01 0.10 0.01 0.11 0.01 0.46 0.00
15 16 -0.53 0.00 0.06 0.01 0.06 0.01 0.35 0.00
16 17 -0.40 0.00 0.03 0.01 0.02 0.01 0.26 0.00
17 18 -0.27 0.00 0.02 0.00 0.02 0.00 0.19 0.00
18 19 -0.15 0.00 0.01 0.00 0.01 0.00 0.14 0.00
19 20 -0.05 0.00 0.01 0.00 0.00 0.00 0.10 0.00
Final results in kJ/mol:
point 0 - 1, DG 0.17 +/- 0.01
point 1 - 2, DG 0.15 +/- 0.01
point 2 - 3, DG 0.11 +/- 0.01
point 3 - 4, DG 0.08 +/- 0.01
point 4 - 5, DG 0.03 +/- 0.00
point 5 - 6, DG -0.01 +/- 0.01
point 6 - 7, DG -0.07 +/- 0.01
point 7 - 8, DG -0.16 +/- 0.01
point 8 - 9, DG -0.26 +/- 0.01
point 9 - 10, DG -0.38 +/- 0.01
point 10 - 11, DG -0.56 +/- 0.01
point 11 - 12, DG -0.81 +/- 0.02
point 12 - 13, DG -1.15 +/- 0.02
point 13 - 14, DG -1.43 +/- 0.03
point 14 - 15, DG -1.48 +/- 0.02
point 15 - 16, DG -1.31 +/- 0.01
point 16 - 17, DG -1.01 +/- 0.01
point 17 - 18, DG -0.68 +/- 0.01
point 18 - 19, DG -0.38 +/- 0.01
point 19 - 20, DG -0.12 +/- 0.00
total 0 - 20, DG -9.26 +/- 0.11
由上可知道本教程 Δ G A B = − 9.11 ± 0.06 k J / m o l = − 2.18 ± 0.01 k c a l / m o l \Delta G_{AB}=-9.11±0.06kJ/mol=-2.18±0.01kcal/mol ΔGAB=−9.11±0.06kJ/mol=−2.18±0.01kcal/mol与教程1中的支撑材料的值 2.24 ± 0.01 k c a l / m o l 2.24±0.01 kcal/mol 2.24±0.01kcal/mol(这里是自由能是正的,是因为原论文是假设不带电的甲烷进入水中与水耦合,与本例甲烷与水解耦相反) 能很好的符合,我们使用的 λ \lambda λ向量的数目大约只有他们的一半。(一般模拟都会进行三次副本模拟,本教程为了演示,只重复一次,和教程1和2的结果有点小误差,是正常范围内的)
以上5和6利用不同的输入数据(dhdl.xvg和md.xvg)文件均会生成以下文件:
├── bar.xvg ;-o选项输出文件
├── barint.xvg ;-oi选项输出文件
├── histogram.xvg ;-oh选项输出文件
└── ... ;更多指定生成文件见gromacs 分析程序说明手册
3.输出文件1: bar.xvg
从bar.xvg开始. 这个输出文件给出了每个 λ \lambda λ区间(即相邻哈密顿量之间)的相对自由能差, 并且看起来应该类似下面的图(使用柱状图绘制, 而没有使用默认的线状图, 为便于查看添加了一些网格线,可以把xvg数据复制到origin自己经过数据处理的到下图:)
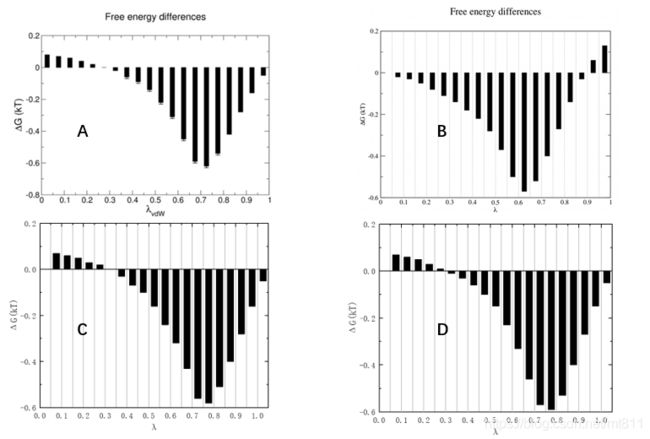
图A: 为教程1中的bar.xvg分析结果图。
图B: 为教程2中的bar.xvg分析结果图,可以看出来它与ACD三个图是相反的,前面说到因为该教程是研究甲烷的溶剂化(甲烷水与发生耦合),而我们设置自由能计算时是用的去耦合(默认输出的图像自由能趋势为图ACD),为了契合主题使用对数据取反( λ = 1 到 λ = 0 \lambda=1到\lambda=0 λ=1到λ=0),可以看出整体数据大小和趋势四张图都是一样。
图C: 为本次教程的配置文件所生成的,使用的calc_lambda_neighbors=-1,与教程1和2的结果基本一致,重复无误。
图D: 为完全重复教程1配置文件,使用的calc_lambda_neighbors=1,与教程1分析结果图A一致,重复无误。
灰色竖线代表去耦合过程中使用的 λ \lambda λ值, 因此, 每一黑色柱代表了相邻 λ \lambda λ值之间的自由能差. 在bar.xvg文件中, 我们首先可以看到模拟中使用了什么calc_lambda_neighbors. 例如在init_lambda_state = 0的模拟中, λ = 0.05 \lambda=0.05 λ=0.05的自由能(最近的临近窗口, 由calc_lambda_neighbors = -1设定)每nstdhdl(100)步计算一次. 这些“外部” λ \lambda λ计算可导致不同 λ \lambda λ值之间的能量发生重叠, 这样就存在相空间之间的重叠, 从而抽样充分. 感兴趣的读者可参看gmx bar说明中引用的Wu和Kofke的论文, 以了解这些概念以及熵值sA和是sB的解释.
关于输出文件barint.xvg和histogram.xvg,与分析bar.xvg类似这里就不在赘述了,详情见教程3
使用alchemical_analysis分析:
1.脚本工具安装:(python2.7+(运行环境)+pymbar(运行依赖包)+其他python依赖包+alchemical_analysis(自由能计算程序))
# 以我的centos为例:
#.1.安装python2.7+
$ sudo yum install python
#2.安装pip
$ sudo wget https://bootstrap.pypa.io/pip/2.7/get-pip.py
$ sudo python2 get-pip.py
#3.安装python依赖包
$ sudo pip2 install numpy
$ sudo pip install six
$ sudo yum install python-devel
$ sudo pip2 install scipy
$ sudo pip install -I pyparsing==2.2.0
$ sudo pip2 install matplotlib
#4安装mbar运行分析依赖包:
$ sudo pip2 install pymbar
#5.到alchemical_analysis目录下安装alchemical_analysis工具
$ sudo python setup.py install
#6.测试是否安装成功:屏幕出选项信息则安装成功
$ alchemical_analysis.py -h
#unbutun下:
#.1.安装python2.7+
$ sudo apt-get install python2.7
#2.安装pip,同上
#3.安装python依赖包,就安装python-dev,需要先安装aptitude
$ sudo pip2 install numpy
$ sudo pip install six
$ sudo yum install python-devel
$ sudo pip2 install scipy
$ sudo apt-get aptitude
$ sudo aptitude install python-dev
#4安装mbar运行分析依赖包:同上
#5.到alchemical_analysis目录下安装alchemical_analysis工具:同上
#6.测试是否安装成功:屏幕出选项信息则安装成功同上
帮助命令详情如下:
-h
显示以下帮助信息和命令选项参数使用细则
-a:指定来自软件包的数据
默认值为Gromacs; 使用: -a <software> (software:Sire,Desmond,AMBER)
例子:使用AMBERS数据进行分析:alchemical_analysis -a AMBER
-b :指定多少分数的能量文件用于统计计算
默认值为1.0; 使用 -b <fraction>
-c: 指定输出基于曲线拟合方法的一致性检查器。
默认值。False. 如图7a;使用 -c <boolean>
-d:指定存储数据文件的目录。
默认值:当前目录; 使用: -d <DATAFILE_DIRECTORY>
-e:指定从后向提取能量数据。
默认值:False; 使用 -e <boolean>
-f:绘制自由能变化在两个方向上的时间函数,时间图中指定的点数。必须提供时间点的数量(一个整数)。
默认值:0 ; 使用:-f <int>
-g:绘制所有方法的每对相邻状态的自由能差评估,包括TI的dH/dlambda曲线。
默认值:False。 -g <boolean>
-i:如果发现非相关样本的数量少于这个数字,则继续进行相关样本的分析。如果给定0,则根本不执行时间序列分析。
默认值:50
-j:自定义的结果文件名前缀。
默认值:result -j <string>
-k: 给出一串用'-'隔开的lambda指数,它们将被从分析中删除。(另一种方法是只让感兴趣的文件存在于目录中)。
默认值:none。 图8分析
-m: 指定列表中的方法来估算自由能量。
默认值。[TI, TI-CUBIC, DEXP, IEXP, BAR, MBAR]。使用:
要添加/删除上述列表中的方法,请提供一个由方法字符串组成的字符串,前面用+/-表示
例如:
-m '-ti_cubic+gdel'将把方法变成 [TI, DEXP, IEXP, BAR, MBAR, GDEL]
-m 'ti_cubic+gdel'将调用 [TI-CUBIC, GDEL]。
-m 'all'将调用所有支持的方法[TI, TI-CUBIC, DEXP, IEXP, GINS, GDEL, BAR, UBAR, RBAR, MBAR]。
-n :用于自相关分析的观测值;可以是 "dhdl_all"(作为所有能量成分的总和)。
默认值:"dhdl"(作为那些正在变化的能量成分的总和)。或 "dE"。
在后一种情况下,使用能量差dE_{i,i+1}(最后一个lambda的dE_{i,i-1})。
-o :脚本产生的输出文件将存放在这个目录中。
默认值:与数据文件目录相同。 使用:-o <OUTPUT_DIRECTORY>
-p :指定要分析数据文件集的文的前缀,
默认值:'dhdl'
-q : 数据文件集的后缀,
默认值:'xvg'
-r :报告自由能的小数点位数。不用担心,这只是文本输出;全精度数据将存储在'results.pickle'中。
默认值:3 ; 使用: -r <int>
-s : 丢弃此指定时间之前的数据作为 "平衡 "数据。单位皮秒。
默认值:0 ps ; 使用: -s <time>
-t : 温度(K),默认值:298 K
-u : 指定报告能量的单位:'kJ'、'kcal'和'kBT'。
默认值:'kJ'; 使用 -u <UNITS>
-v: 冗长选项。
默认值。False.
-w:打印并绘制重叠矩阵。 图7b
默认值:False。
报错处理:#O = MBAR.computeOverlap()[2]改成
O = MBAR.computeOverlap()['matrix']
-x:不检查WL权重是否平衡。
不需要日志文件作为附带输入。
-y:BAR和MBAR能量估计的收敛标准。
默认值:1e-10。
-z: 初始的MBAR自由能量猜测;可以是 "BAR "或 "零"。
默认值:'bar'。
使用方法,在数据集合文件中:
alchemical_analysis -p dhdl -u kcal
或者
alchemical_analysis -t 300 -s 0 -u kcal (-w -g) :坑
注意下-w,-g可以不要,会报错
2.使用 alchemical_analysis工具分析dhdl.xvg系列数据:(本教程分析方法)
#1.calc-lambda-neighbors = -1,使用单独生成的dhdl.xvg文件的(从0-20)
#默认输出基于TI自由能计算方法(TI,TI-CUBIC),基于FEP(DEXP.IEXP,BAR,MBAR),6种计算结果
#在含有dhdl.xvg文件夹输入下列命令
$ alchemical_analysis -p dhdl -u kJ
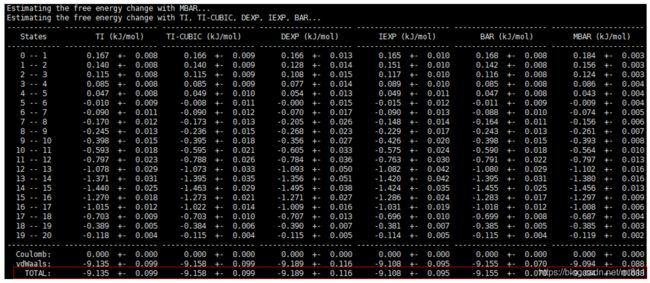
#输出全部基于FEP自由能计算方法的结果:共8种:
$ alchemical_analysis -p dhdl -t 300 -u kJ -m 'dexp+iexp+gins+gdel+ubar+rbar+bar+mbar'
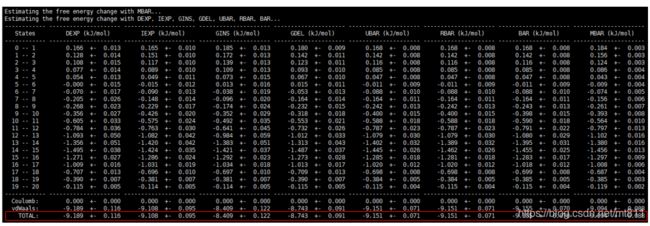
从以上两张图可以发现使用alchemical_analysis计算的自由能的结果同gmx bar分析出自由能结果是一致的均为程 Δ G A B = − 9.11 ± 0.06 k J / m o l \Delta G_{AB}=-9.11±0.06kJ/mol ΔGAB=−9.11±0.06kJ/mol
(1)绘制$\Delta G 与 与 与\lambda$ 关系图(FEP:同bar.xvg图)
#使用 -g 选项绘制-m 选项的指定的方法的图像,fep_out是我自己指定的文件夹用来保存输出的文件
$ alchemical_analysis -p dhdl -t 300 -u kBT -m 'dexp+iexp+gins+gdel+ubar+rbar+bar+mbar' -g -o fep_out
#会生成dF_state.pdf;dF_state_long.pdf;文件,文件图像在PDF文件中。

可以看出来与gmx bar 分析出来的bar.xvg作出来的图基本是一致的。
(2)绘制正向和反向的自由能随着模拟时间变化的函数图:本例是(forward,去耦合,那么反向就是去耦合)
#使用 -f选项,会生成dF_t.pdf文件
$ alchemical_analysis -p dhdl -u kBT -f 21 -o fep_out

(3)绘制相空间重叠矩阵
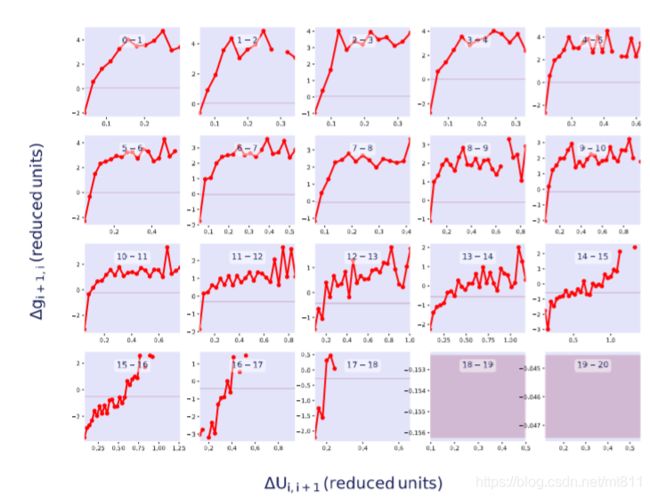
#使用 -c选项,会生成O_MBAR.pdf文件
$ alchemical_analysis -p dhdl -u kBT -c -o fep_out

(4)绘制重叠分布法:
#使用 -c选项,会生成cfm.pdf文件
$ alchemical_analysis -p dhdl -u kBT -w -o fep_out
3.使用 alchemical_analysis工具分析md.xvg系列数据:(教程2模拟生成结果,用此工具来分析)
#calc-lambda-neighbors = 1,使用md.xvg(0-20),md.xvg里面包括dhdl.xvg的数据会发现没有了mbar选项
#与mbar相关图就没法做了如O_MBAR.pdf
$ alchemical_analysis -p md -u kcal
总结
alchemical_analysis是一款强大的自由能计算后处理分析程序,支持amber,gromacs,sire分子模拟软件包自由能计算的结果的分析,集成了matplotlib,能绘制丰富的与自由能相关图像。本程序是根据自由能专题2:计算与分析指南中自由能分析最佳做法来进行自动化处理分析的,主要有是基于TI和FEP两大自由能计算方法进行分析,并提供了大量对自由能计算结果的收敛性分析,以用来评估自由能计算结果的质量,通过与原生分析程序gmx bar(gromacs自带的)对比,可以发现,该程序不仅计算结果准确,而且还能基于同一输入文件,进行一站式的使用多种基于TI和FEP后处理方法进行自动化分析,和原生工具无需进行分析前后的数据处理与图形绘制。
3.4 TI:计算甲烷在水中的溶剂化自由能
这一部分与在gromacs中与3.3中FEP自由能计算共用一套代码,FEP生成的结果同样可以适用于基于TI自由能计算方法的后处理分析,因为前面说到,使用gromcas自由能计算代码可生成dhdl.xvg文件(含 ⟨ ∂ H ∂ λ ⟩ \langle \frac{ \partial H} { \partial \lambda} \rangle ⟨∂λ∂H⟩和 Δ H \Delta H ΔH值)或包含dhdl.xvg文件数据的md.xvg文件,其中 Δ H \Delta H ΔH值用于基于FEP的自由能分析,而 ⟨ ∂ H ∂ λ ⟩ \langle \frac{ \partial H} { \partial \lambda} \rangle ⟨∂λ∂H⟩用于基于TI的自由能分析。
分析
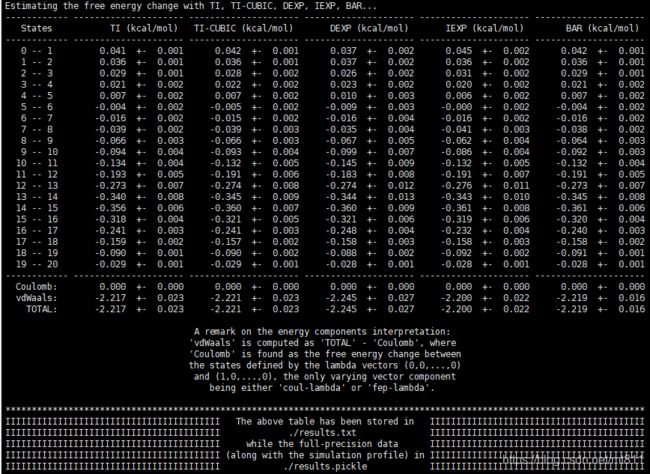
其他的均与3.3中的分析相同,就以下为TI分析图像
(1)绘制 ⟨ ∂ H ∂ λ ⟩ \langle \frac{ \partial H} { \partial \lambda} \rangle ⟨∂λ∂H⟩与 λ \lambda λ关系图像(TI),如图所示我们在模拟是仅开关了VDW相互作用,所下图仅显示vdw
$ alchemical_analysis -p dhdl -t 300 -u kBT -m 'ti' -g -o fep_out
#会生成文件,文件图像在PDF文件中。

3.5.(Jarzynski Equality)非平衡近似
1.以上我们已经使用TI和FEP两种方法计算自由能,它们两者都是对称的,当 λ \lambda λ间隔足够的小,相应的 λ \lambda λ系综间有重叠时,这些方法能够给出精确的自由能估计值,而, 由于采样时间的限制, 实际情况很少这样. 与平衡方法相反, Jarzynski方法用于远离平衡的体系. 尽管考虑到我们的限制, 这听起来不错, 但由于平均问题, 需要非常多的模拟才能得到精确的自由能估计值. 最后的结果就是你并不能节省多少模拟时间. 这样, 具体使用哪种方法依赖于你要研究的问题, 要由你来决定使用哪种方法。
设置
同以FEP和TI自由能计算代码一样,主要修改自由能代码的以下部分:
delta_lambda = 0.0001 ; 每步的增量, 慢增长方法取非零值
其他部分都是一样的
分析
1.执行完所有模拟后, 可以使用anlyze程序计算所有模拟的非可逆功:
#分析一系列的dhdl.xvg文件
$ gmx analyze -f dhdl*.xvg -integrate
注意, x值是以ps为单位的时间, 从0到20. 因此你需要将所得的功除以20使得 λλ 处于0到1之间, 间隔正确. 得到这些功后, 你可以使用你熟悉的数据分析程序创建一个直方图, 并试着将其拟合为高斯函数, 使用Jarzynski 方程(指数平均)来计算可逆功:
e x p [ − β Δ F ] = 1 n s i m ∑ n s i m e x p [ − β ∫ t i t f ∂ V ∂ λ d V d t d t ] exp[-\beta \Delta F]=\frac{1}{n_{sim}}\sum_{n_{sim}}exp [-\beta\int_{t_i}^{t_f}\frac{\partial V}{\partial \lambda}\frac{d V}{dt}dt] exp[−βΔF]=nsim1nsim∑exp[−β∫titf∂λ∂VdtdVdt]
2.也能使用TI和FEP中的分析方法
3.6 MM/PBSA
1.使用gmx_mm/pbsa脚本计算, MM-PBSA(Molecular Mechanics-Poisson Bolzmann Surface Area, 分子力学泊松玻尔兹曼表面积)是一种对MD轨迹进行后处理以估计结合自由能的方法, 计算时会将溶剂视为均匀的连续介质, 并基于力场和隐式的连续介质模型, 对平衡轨迹中的许多帧进行平均, 以考虑温度的影响,以下教程已经很详细了,这里不在赘述:
1.MM/PBSA计算原理文章1
2.GROMACS教程:使用GROMACS计算MM-PBSA结合自由能
2.gmx_mmpbsa使用说明
3.gmx_mmpbsa脚本更新:屏蔽效应与熵贡献
安装与使用
主要工具:gromacs + gmx_mm/pbsa(脚本) + APBS,gromacs在这里的安装就不在赘述了
1.APBS 在unbutun下能直接安装:
$ sudo apt install apbs
2.gmx_mm/pbsa脚本:来自哲科文李老师
echo -e "\
>>>>>>>>>>>>>>>> gmx_mmpbsa <<<<<<<<<<<<<<<<
>>>>>>>>>>>>>>>> Jicun Li <<<<<<<<<<<<<<<<
>>>>>>>>>> 2020-11-15 10:11:59 <<<<<<<<<\n
>> Usage: gmx_mmpbsa -f *.xtc -s *.tpr -n *.ndx
-com COMPLEX -pro PROTEIN {-lig }\n
>> Default: gmx_mmpbsa -f traj.xtc -s topol.tpr -n index.ndx
-com Complex -pro Protein -lig Ligand
>> Option:
f: trajectory file
s: topology file
n: index file
com: index group name of complex
pro: index group name of protein
lig: index group name of ligand, can be ignored using none
change other settings in the script directly
>> Log:
TODO: Opt before MM-PBSA
TODO: CAS
TODO: parallel APBS, focus
2020-11-15: fix bug for resID >=1000, for awk 3.x gsub /\s/
2020-06-02: fix bug of withLig
2020-06-01: fix bug of -skip
2020-05-27: fix bug for sdie
2020-05-26: fix bug for RES name
2020-04-03: use C6, C12 directly
2020-01-08: support protein only
2019-12-24: fix bug for small time step
2019-12-10: fix bug for OPLS force field
2019-11-17: fix bug for c6, c12 of old version tpr
2019-11-06: apbs FILE.apbs &> FILE.out
on ubuntu 18.04 may not work, then delete &
2019-11-03: fix bug for time stamp
2019-09-19: push to gmxtool
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>\n"
################################################################################
# 设置运行环境, 计算参数
# setting up environmets and parameters
################################################################################
trj=traj.xtc # 轨迹文件 trajectory file
tpr=topol.tpr # tpr文件 tpr file
ndx=index.ndx # 索引文件 index file
com=Complex # 复合物索引组 index group name of complex
pro=Protein # 蛋白索引组 index group name of protein
lig=Ligand # 配体索引组 index group name of ligand
step=1 # 从第几步开始运行 step number to run
# 1. 预处理轨迹: 复合物完整化, 团簇化, 居中叠合, 然后生成pdb文件
# pre-processe trajectory, whole, cluster, center, fit, then generate pdb file
# 2. 获取每个原子的电荷, 半径, LJ参数, 然后生成qrv文件
# abstract atomic parameters, charge, radius, C6/C12, then generate qrv file
# 3. MM-PBSA计算: pdb->pqr, 输出apbs, 计算MM, APBS
# run MM-PBSA, pdb->pqr, apbs, then calculate MM, PB, SA
gmx='gmx' # /path/to/GMX/bin/gmx_mpi
dump="$gmx dump" # gmx dump
trjconv="$gmx trjconv -dt 1000" # gmx trjconv, use -b -e -dt, NOT -skip
#apbs='/path/APBS/bin/apbs' # APBS(Linux)
apbs='c:/apbs1.5/bin/apbs.exe' # APBS(Windows), USE "/", NOT "\"
export MCSH_HOME=/dev/null # APBS io.mc
pid=pid # 输出文件($$可免重复) prefix of the output files($$)
scr=_$pid.scr # 屏幕输出文件 file to save the message from the screen
qrv=_$pid.qrv # 电荷/半径/VDW参数文件 to save charge/radius/vdw parmeters
radType=1 # 原子半径类型 radius of atoms (0:ff; 1:mBondi; 2:Bondi)
radLJ0=1.2 # 用于LJ参数原子的默认半径(A, 主要为H) radius when LJ=0 (H)
meshType=0 # 网格大小 mesh (0:global 1:local)
gridType=1 # 格点大小 grid (0:GMXPBSA 1:psize)
cfac=3 # 分子尺寸到粗略格点的放大系数
# Factor to expand mol-dim to get coarse grid dim
fadd=10 # 分子尺寸到细密格点的增加值(A)
# Amount added to mol-dim to get fine grid dim (A)
df=.5 # 细密格点间距(A) The desired fine mesh spacing (A)
# 极性计算设置(Polar)
PBEset='
temp 298.15 # 温度
pdie 2 # 溶质介电常数
sdie 78.54 # 溶剂介电常数, 真空1, 水78.54
lpbe # PB方程求解方法, lpbe(线性), npbe(非线性), smbpe(大小修正)
bcfl mdh # 粗略格点PB方程的边界条件, zero, sdh/mdh(single/multiple Debye-Huckel), focus, map
srfm smol # 构建介质和离子边界的模型, mol(分子表面), smol(平滑分子表面), spl2/4(三次样条/7阶多项式)
chgm spl4 # 电荷映射到格点的方法, spl0/2/4, 三线性插值, 立方/四次B样条离散
swin 0.3 # 立方样条的窗口值, 仅用于 srfm=spl2/4
srad 1.4 # 溶剂探测半径
sdens 10 # 表面密度, 每A^2的格点数, (srad=0)或(srfm=spl2/4)时不使用
ion 1 0.15 0.95 # 阳离子的电荷, 浓度, 半径
ion -1 0.15 1.81 # 阴离子
calcforce no
calcenergy comps'
# 非极性计算设置(Apolar/Non-polar)
PBAset='
temp 298.15 # 温度
srfm sacc # 构建溶剂相关表面或体积的模型
swin 0.3 # 立方样条窗口(A), 用于定义样条表面
# SASA
srad 1.4 # 探测半径(A)
gamma 1 # 表面张力(kJ/mol-A^2)
#gamma const 0.027 0 # 表面张力, 常数
#gamma const 0.0226778 3.84928 # 表面张力, 常数
press 0 # 压力(kJ/mol-A^3)
bconc 0 # 溶剂本体密度(A^3)
sdens 10
dpos 0.2
grid 0.1 0.1 0.1
# SAV
#srad 1.29 # SAV探测半径(A)
#press 0.234304 # 压力(kJ/mol-A^3)
# WCA
#srad 1.25 # 探测半径(A)
#sdens 200 # 表面的格点密度(1/A)
#dpos 0.05 # 表面积导数的计算步长
#bconc 0.033428 # 溶剂本体密度(A^3)
#grid 0.45 0.45 0.45 # 算体积分时的格点间距(A)
calcforce no
calcenergy total'
################################################################################
# 检查 gmx, apbs 是否可以运行
# check gmx, apbs
################################################################################
str=$($gmx --version | grep -i "GROMACS version")
[[ -z "$str" ]] && { echo -e "!!! WARNING !!! GROMACS NOT available !\n"; }
str=$($apbs --version | grep -i "Poisson-Boltzmann")
[[ -z "$str" ]] && { echo -e "!!! WARNING !!! APBS NOT available !\n"; }
################################################################################
# 解析命令行参数
# parse command line options
################################################################################
opt=($*); N=${#opt[@]}
for((i=0; i<N; i++)); do
arg=${opt[$i]}; j=$((i+1)); val=${opt[$j]}
[[ $arg =~ -f ]] && { trj=$val; }
[[ $arg =~ -s ]] && { tpr=$val; }
[[ $arg =~ -n ]] && { ndx=$val; }
[[ $arg =~ -com ]] && { com=$val; }
[[ $arg =~ -pro ]] && { pro=$val; }
[[ $arg =~ -lig ]] && { lig=$val; }
done
withLig=1; [[ $lig =~ none ]] && { withLig=0; com=$pro; lig=$pro; }
################################################################################
# 检查文件
# check files
################################################################################
[[ ! -f "$trj" ]] && { echo -e "!!! ERROR !!! trajectory File NOT Exist !\n"; exit; }
[[ ! -f "$tpr" ]] && { echo -e "!!! ERROR !!! topology File NOT Exist !\n"; exit; }
[[ ! -f "$ndx" ]] && { echo -e "!!! ERROR !!! index File NOT Exist !\n"; exit; }
str=$(grep $com "$ndx"); [[ -z "$str" ]] && { echo -e "!!! ERROR !!! $com NOT in $ndx !\n"; exit; }
str=$(grep $pro "$ndx"); [[ -z "$str" ]] && { echo -e "!!! ERROR !!! $pro NOT in $ndx !\n"; exit; }
str=$(grep $lig "$ndx"); [[ -z "$str" && $withLig -eq 1 ]] && { echo -e "!!! ERROR !!! $lig NOT in $ndx !\n"; exit; }
if [[ $step -le 1 ]]; then
################################################################################
# 1. 预处理轨迹: 复合物完整化, 团簇化, 居中叠合, 然后生成pdb文件
# 请检查pdb文件确保构型PBC处理正确
# 1. pre-processe trajectory, whole, cluster, center, fit, then generate pdb file
################################################################################
trjwho=$pid~who; trjcnt=$pid~cnt; trjcls=$pid~cls
echo $com | $trjconv -s $tpr -n $ndx -f $trj -o $trjwho.xtc &> $scr -pbc whole
echo -e "$lig\n$com" | $trjconv -s $tpr -n $ndx -f $trjwho -o _$pid.pdb &>>$scr -pbc mol -center
# usful for single protein and ligand
#echo $com | $trjconv -s $tpr -n $ndx -f $trj -o $trjwho.xtc &> $scr -pbc whole
#echo -e "$lig\n$com" | $trjconv -s $tpr -n $ndx -f $trjwho -o $trjcnt.xtc &>>$scr -pbc mol -center
#echo -e "$com\n$com" | $trjconv -s $tpr -n $ndx -f $trjcnt -o $trjcls.xtc &>>$scr -pbc cluster
#echo -e "$lig\n$com" | $trjconv -s $tpr -n $ndx -f $trjcls -o _$pid.pdb &>>$scr -fit rot+trans
fi; if [[ $step -le 2 ]]; then
################################################################################
# 2. 获取每个原子的电荷, 半径, LJ参数, 然后生成qrv文件
# 2. abstract atomic parameters, charge, radius, C6/C12, then generate qrv file
# feel free to change radius with radType
# radType=0: radius from C6/C12, or radLJ0 if either C6 or C12 is zero
# radType=1: mBondi
# radType=2: Bondi
################################################################################
$dump -quiet -s $tpr 2>>$scr \
| awk >$qrv -v ndx=$ndx -v pro=$pro -v lig=$lig -v withLig=$withLig \
-v radType=$radType -v radLJ0=$radLJ0 '
BEGIN { RS="["
print pro, lig
while(getline < ndx) {
gsub(" ","", $1); gsub("\t","", $1)
if($1==pro) for(i=3; i<=NF; i++) ndxPro[$i+0]++
if($1==pro"]") for(i=2; i<=NF; i++) ndxPro[$i+0]++
if(withLig) {
if($1==lig) for(i=3; i<=NF; i++) ndxLig[$i+0]++
if($1==lig"]") for(i=2; i<=NF; i++) ndxLig[$i+0]++
}
}
RS="\r?\n"
nres=0
}
/#molblock/ { Ntyp=$3 }
/moltype.+=/ { Imol=$3; getline; Nmol[Imol]=$3 }
/ffparams:/ {
getline Atyp; sub(/.+=/, "", Atyp); Atyp += 0
print Atyp
getline
for(i=0; i$0 ; sub(".*c6 *= *", "", C6); sub(",.*", "", C6);
C12=$0; sub(".*c12 *= *", "", C12); sub(",.*", "", C12);
printf " %s %s", C6, C12
if(j==i) {
sigma[i]=0; epsilon[i]=0
Rad[i]=radLJ0
if(C6*C12!=0) {
sigma[i]=10*(C12/C6)^(1./6) # 转换单位为A
epsilon[i]=C6^2/(4*C12)
Rad[i]=.5*sigma[i] # sigma为直径
}
}
}
print ""
}
}
/moltype.+\(/ { Imol=$0; gsub(/[^0-9]/,"",Imol)
getline txt; sub(/.*=/,"",txt); gsub(" ","_",txt)
Name[Imol]=txt
getline; getline txt; gsub(/[^0-9]/,"",txt); Natm[Imol]=txt+0
for(i=0; i$0; idx=$3; resID[Imol, i]=$(NF-2)+1+nres
sub(",", "", idx); idx += 0;
Catm[Imol, i]=idx
Ratm[Imol, i]=Rad[idx]
Satm[Imol, i]=sigma[idx]
Eatm[Imol, i]=epsilon[idx]
sub(/.+q=/, "", txt); sub(/,.+/, "", txt); Qatm[Imol, i]=txt
}
getline
for(i=0; i$0); sub(/".*/,"",$0);
resName[nres]=sprintf("%d%s", nres, $0)
}
END {
Ntot=0; Nidx=0
for(i=0; i0) radi=getRadi(Tatm[i, j], radType)
printf "%6d %9.5f %9.6f %6d %9.6f %9.6f %6d %s %s %-6s ", \
Nidx, Qatm[i,j], radi, Catm[i,j], Satm[i,j], Eatm[i,j], \
Ntot, Name[i]"-"n+1"."j+1, \
resName[resID[i,j]], Tatm[i, j]
if(Ntot in ndxPro) print "Pro"
if(Ntot in ndxLig) print "Lig"
}
}
}
}
}
function getRadi(tag, radType) {
radBondi["O" ]= 1.50; if(radType==2) radBondi["O" ]= 1.52
radBondi["S" ]= 1.80; if(radType==2) radBondi["S" ]= 1.83
radBondi["P" ]= 1.85; if(radType==2) radBondi["P" ]= 1.80
radBondi["I" ]= 1.98; if(radType==2) radBondi["I" ]= 2.06
radBondi["BR"]= 1.85; if(radType==2) radBondi["BR"]= 1.92
radBondi["N" ]= 1.55
radBondi["F" ]= 1.47
radBondi["CL"]= 1.77
radBondi["C" ]= 1.70; radBondi["H" ]= 1.20
radBondi["C*"]= 1.77; radBondi["H4"]= 1.00
radBondi["CA"]= 1.77; radBondi["H5"]= 1.00
radBondi["CB"]= 1.77; radBondi["HA"]= 1.00
radBondi["CC"]= 1.77; radBondi["HC"]= 1.30
radBondi["CD"]= 1.77; radBondi["HN"]= 1.30
radBondi["CN"]= 1.77; radBondi["HP"]= 1.30
radBondi["CR"]= 1.77; radBondi["HO"]= 0.80
radBondi["CV"]= 1.77; radBondi["HS"]= 0.80
radBondi["CW"]= 1.77;
tag=toupper(tag)
if(length(tag)>=2) {
if(!radBondi[substr(tag,1,2)]) return radBondi[substr(tag,1,1)]
else return radBondi[substr(tag,1,2)]
}
return radBondi[tag]
}
'
fi; if [[ $step -le 3 ]]; then
################################################################################
# 3. MM-PBSA计算: pdb->pqr, 输出apbs, 计算MM, APBS
# 3. run MM-PBSA, pdb->pqr, apbs, then calculate MM, PB, SA
################################################################################
dt=$(awk '/t=/{n++;sub(/.*t=/,"");sub(/step=.*/,"");t[n]=$0;if(n==2){print t[n]-t[1];exit}}' _$pid.pdb)
awk -v pid=_$pid -v qrv=$qrv -v apbs="$apbs" \
-v ff=$ff -v PBEset="$PBEset" -v PBAset="$PBAset" \
-v meshType=$meshType -v gridType=$gridType -v gmem=$gmem \
-v fadd=$fadd -v cfac=$cfac -v df=$df -v dt="$dt" \
-v withLig=$withLig -v RS="\r?\n" '
BEGIN {
getline < qrv
getline Atyp < qrv
for(i=0; i$(2+2*j) ; C12[i,j]=$(3+2*j) }
}
while(getline < qrv) {
Qatm[$1]=$2; Ratm[$1]=$3; Catm[$1]=$4
Satm[$1]=$5; Eatm[$1]=$6
if($NF=="Pro") { Npro++; if(Npro==1) Ipro=$1
ndxPro[$1]++; resPro[Npro]="P~"$(NF-2)
}
if($NF=="Lig") { Nlig++; if(Nlig==1) Ilig=$1
ndxLig[$1]++; resLig[Nlig]="L~"$(NF-2)
}
}
close(qrv)
Ncom=Npro+Nlig
PBEset0=PBEset; sub(/sdie +[0-9]*\.*[0-9]*/, "sdie 1", PBEset0)
txt=PBEset; sub(/.*pdie +/, "", txt);
sub(/\n.*/, "", txt); split(txt, arr)
pdie=arr[1]
txt=PBAset; sub(/.*#gamma +con[a-zA-Z]+/, "", txt);
sub(/\n.*/, "", txt); split(txt, arr)
gamma=arr[1]; const=arr[2]
MAXPOS=1E9
minX= MAXPOS; maxX=-MAXPOS;
minY= MAXPOS; maxY=-MAXPOS;
minZ= MAXPOS; maxZ=-MAXPOS
fmt=sprintf("%.9f",dt/1E3)
sub(/0*$/,"",fmt);sub(/.*\./,"",fmt)
fmt="~%."length(fmt)"fns"
}
/REMARK/ {next}
/TITLE/ {Fout=FILENAME
txt=$0; sub(/.*t= */,"",txt); sub(/ .*/,"",txt)
txt=sprintf(fmt, txt/1E3);
sub(".pdb", txt, Fout)
Nfrm++; n=0
Fname[Nfrm]=Fout
minXpro[Nfrm]= MAXPOS; minXlig[Nfrm]= MAXPOS;
minYpro[Nfrm]= MAXPOS; minYlig[Nfrm]= MAXPOS;
minZpro[Nfrm]= MAXPOS; minZlig[Nfrm]= MAXPOS
maxXpro[Nfrm]=-MAXPOS; maxXlig[Nfrm]=-MAXPOS
maxYpro[Nfrm]=-MAXPOS; maxYlig[Nfrm]=-MAXPOS
maxZpro[Nfrm]=-MAXPOS; maxZlig[Nfrm]=-MAXPOS
}
/^ATOM/ {
ATOM=substr($0,1,6)
INDX=substr($0,7,5)+0
NAME=substr($0,13,4)
RES =substr($0,18,3)
CHN =substr($0,22,1); if(CHN=" ") CHN="A"
NUM =substr($0,23,4)
X =substr($0,31,8); X += 0
Y =substr($0,39,8); Y += 0
Z =substr($0,47,8); Z += 0
r=Ratm[INDX]
txt=sprintf("%-6s%5d %-4s %3s %s%4d %8.3f %8.3f %8.3f %12.6f %12.6f", \
ATOM, INDX, NAME, RES, CHN, NUM, X, Y, Z, Qatm[INDX], r)
if(INDX in ndxPro) {
print txt > Fout"_pro.pqr"
minXpro[Nfrm]=min(minXpro[Nfrm], X-r); maxXpro[Nfrm]=max(maxXpro[Nfrm], X+r)
minYpro[Nfrm]=min(minYpro[Nfrm], Y-r); maxYpro[Nfrm]=max(maxYpro[Nfrm], Y+r)
minZpro[Nfrm]=min(minZpro[Nfrm], Z-r); maxZpro[Nfrm]=max(maxZpro[Nfrm], Z+r)
}
if(withLig) {
print txt > Fout"_com.pqr"
if(INDX in ndxLig) {
print txt > Fout"_lig.pqr"
minXlig[Nfrm]=min(minXlig[Nfrm], X-r); maxXlig[Nfrm]=max(maxXlig[Nfrm], X+r)
minYlig[Nfrm]=min(minYlig[Nfrm], Y-r); maxYlig[Nfrm]=max(maxYlig[Nfrm], Y+r)
minZlig[Nfrm]=min(minZlig[Nfrm], Z-r); maxZlig[Nfrm]=max(maxZlig[Nfrm], Z+r)
}
}
minXcom[Nfrm]=min(minXpro[Nfrm], minXlig[Nfrm]); maxXcom[Nfrm]=max(maxXpro[Nfrm], maxXlig[Nfrm])
minYcom[Nfrm]=min(minYpro[Nfrm], minYlig[Nfrm]); maxYcom[Nfrm]=max(maxYpro[Nfrm], maxYlig[Nfrm])
minZcom[Nfrm]=min(minZpro[Nfrm], minZlig[Nfrm]); maxZcom[Nfrm]=max(maxZpro[Nfrm], maxZlig[Nfrm])
minX=min(minX, minXcom[Nfrm]); maxX=max(maxX, maxXcom[Nfrm])
minY=min(minY, minYcom[Nfrm]); maxY=max(maxY, maxYcom[Nfrm])
minZ=min(minZ, minZcom[Nfrm]); maxZ=max(maxZ, maxZcom[Nfrm])
next
}
END{
kJcou=1389.35457520287
Rcut=1E10 # large enough
for(i=1; i<=Npro; i++) dE[resPro[i]]=0
for(i=1; i<=Nlig; i++) dE[resLig[i]]=0
Nres=asorti(dE, Tres)
txt=" #Frame "
for(i=1; i<=Nres; i++) {
ii=Tres[i]; sub(/~0+/, "~", ii)
txt = txt""sprintf("%12s", ii)
}
if(withLig) {
print txt > pid"~resMM.dat"
print txt > pid"~resMM_COU.dat"
print txt > pid"~resMM_VDW.dat"
print txt > pid"~res_MMPBSA.dat"
}
print txt > pid"~resPBSA.dat"
print txt > pid"~resPBSA_PB.dat"
print txt > pid"~resPBSA_SA.dat"
print " #Frame Binding MM PB SA "\
"| COU VDW | PBcom PBpro PBlig "\
"| SAcom SApro SAlig" >> pid"~MMPBSA.dat"
for(fr=1; fr<=Nfrm; fr++) {
Fout=Fname[fr]
print "running for Frame "fr": "Fout
txt=Fout"_pro.pqr"; if(withLig) txt=Fout"_com.pqr";
close(txt)
n=0;
while(getline < txt) { n++;
type[n]=$3; res[n]=$4;
x[n]=$(NF-4); y[n]=$(NF-3); z[n]=$(NF-2)
resID[n]=$(NF-5); gsub(/[A-Z]+/, "", resID[n])
}
close(txt)
# MM
if(withLig) {
for(i=1; i<=Npro; i++) { dEcou[resPro[i]]=0; dEvdw[resPro[i]]=0 }
for(i=1; i<=Nlig; i++) { dEcou[resLig[i]]=0; dEvdw[resLig[i]]=0 }
for(i=1; i<=Npro; i++) {
ii=i+Ipro-1
qi=Qatm[ii]; ci=Catm[ii]; si=Satm[ii]; ei=Eatm[ii]
xi=x[ii]; yi=y[ii]; zi=z[ii]
for(j=1; j<=Nlig; j++) {
jj=j+Ilig-1; cj=Catm[jj]
r=sqrt( (xi-x[jj])^2+(yi-y[jj])^2+(zi-z[jj])^2 )
if(r Fout".apbs"
else print "read\n" \
" mol pqr "Fout"_pro.pqr\n" \
"end\n\n" > Fout".apbs"
if(meshType==0) { # GMXPBSA
if(withLig) print \
dimAPBS(Fout"_com", 1, minX, maxX, minY, maxY, minZ, maxZ), \
dimAPBS(Fout"_pro", 2, minX, maxX, minY, maxY, minZ, maxZ), \
dimAPBS(Fout"_lig", 3, minX, maxX, minY, maxY, minZ, maxZ) > Fout".apbs"
else print \
dimAPBS(Fout"_pro", 1, minX, maxX, minY, maxY, minZ, maxZ) > Fout".apbs"
} else if(meshType==1) { # g_mmpbsa
if(withLig) print \
dimAPBS(Fout"_com", 1, minXcom[fr], maxXcom[fr], minYcom[fr], maxYcom[fr], minZcom[fr], maxZcom[fr]), \
dimAPBS(Fout"_pro", 2, minXpro[fr], maxXpro[fr], minYpro[fr], maxYpro[fr], minZpro[fr], maxZpro[fr]), \
dimAPBS(Fout"_lig", 3, minXlig[fr], maxXlig[fr], minYlig[fr], maxYlig[fr], minZlig[fr], maxZlig[fr]) > Fout".apbs"
else print \
dimAPBS(Fout"_pro", 1, minXpro[fr], maxXpro[fr], minYpro[fr], maxYpro[fr], minZpro[fr], maxZpro[fr]) > Fout".apbs"
}
cmd=apbs" "Fout".apbs > "Fout".out 2>&1";
system(cmd); close(cmd)
txt=Fout".out";
while(getline < txt ) {
if(index($0, "CALCULATION #")) {
if(index($0, "("Fout"_com")) { t=1; n=Ncom }
if(index($0, "("Fout"_pro")) { t=2; n=Npro }
if(index($0, "("Fout"_lig")) { t=3; n=Nlig }
if(index($0, "~VAC)")) t += 10
if(index($0, "~SAS)")) t += 20
while(getline < txt) {
if(t<20 && index($0, "Per-atom energies:") \
|| t>20 && index($0, "Solvent Accessible Surface Area")) break
}
for(i=1; i<=n; i++) {
getline $3; else r=$NF
if(t<10) Esol[t%10, i]=r
else if(t<20) Evac[t%10, i]=r
else if(t<30) Esas[t%10, i]=gamma*r+const/n
}
}
}
close(txt)
PBcom=0; SAcom=0;
PBpro=0; SApro=0;
PBlig=0; SAlig=0;
for(i=1; i<=Ncom; i++) { Esol[1,i] -= Evac[1,i]; PBcom += Esol[1,i]; SAcom += Esas[1,i] }
for(i=1; i<=Npro; i++) { Esol[2,i] -= Evac[2,i]; PBpro += Esol[2,i]; SApro += Esas[2,i] }
for(i=1; i<=Nlig; i++) { Esol[3,i] -= Evac[3,i]; PBlig += Esol[3,i]; SAlig += Esas[3,i] }
for(i=1; i<=Npro; i++) { PBres[resPro[i]]=0; SAres[resPro[i]]=0 }
for(i=1; i<=Nlig; i++) { PBres[resLig[i]]=0; SAres[resLig[i]]=0 }
for(i=1; i<=Npro; i++) {
PBres[resPro[i]] += Esol[1, Ipro+i-1]-Esol[2, i]
SAres[resPro[i]] += Esas[1, Ipro+i-1]-Esas[2, i]
}
for(i=1; i<=Nlig; i++) {
PBres[resLig[i]] += Esol[1, Ilig+i-1]-Esol[3, i]
SAres[resLig[i]] += Esas[1, Ilig+i-1]-Esas[3, i]
}
preK=-1; if(withLig) preK=1
printf "%-12s %9.3f %9.3f %9.3f %9.3f | %9.3f %9.3f | %12.3f %12.3f %12.3f | %9.3f %9.3f %9.3f\n", \
Fout, preK*(Ecou+Evdw+PBcom-PBpro-PBlig+SAcom-SApro-SAlig), \
Ecou+Evdw, preK*(PBcom-PBpro-PBlig), preK*(SAcom-SApro-SAlig), \
Ecou, Evdw, PBcom, PBpro, PBlig, SAcom, SApro, SAlig >> pid"~MMPBSA.dat"
fmt="%s%12.3f%s"
for(i=1; i<=Nres; i++) {
ii=""; if(i==1) ii=sprintf("%-12s", Fout)
txt=""; if(i==Nres) txt="\n"
if(withLig) {
printf fmt, ii, dEcou[Tres[i]], txt > pid"~resMM_COU.dat"
printf fmt, ii, dEvdw[Tres[i]], txt > pid"~resMM_VDW.dat"
printf fmt, ii, dEcou[Tres[i]]+dEvdw[Tres[i]], txt > pid"~resMM.dat"
printf fmt, ii, dEcou[Tres[i]]+dEvdw[Tres[i]] \
+PBres[Tres[i]]+SAres[Tres[i]], txt > pid"~res_MMPBSA.dat"
}
printf fmt, ii, preK*(PBres[Tres[i]]), txt > pid"~resPBSA_PB.dat"
printf fmt, ii, preK*(SAres[Tres[i]]), txt > pid"~resPBSA_SA.dat"
printf fmt, ii, preK*(PBres[Tres[i]]+SAres[Tres[i]]), txt > pid"~resPBSA.dat"
}
fmt="%s%6.1f%6.1f\n"
for(i=1; i<=Npro; i++) {
ii=Ipro+i-1
txt=sprintf("%-6s%5d %-4s %3s A%4d %8.3f%8.3f%8.3f", \
"ATOM", ii, type[ii], res[ii], resID[ii], x[ii], y[ii], z[ii])
if(withLig) {
printf fmt, txt, dEcou[resPro[i]], dEvdw[resPro[i]] > Fout"~COU+VDW.pdb"
printf fmt, txt, dEcou[resPro[i]]+dEvdw[resPro[i]], \
PBres[resPro[i]]+SAres[resPro[i]] > Fout"~res_MM+PBSA.pdb"
}
printf fmt, txt, preK*PBres[resPro[i]], preK*SAres[resPro[i]] > Fout"~PB+SA.pdb"
printf fmt, txt, 0, dEcou[resPro[i]]+dEvdw[resPro[i]] \
+preK*(PBres[resPro[i]]+SAres[resPro[i]]) > Fout"~res_MMPBSA.pdb"
}
for(i=1; i<=Nlig; i++) {
ii=Ilig+i-1
txt=sprintf("%-6s%5d %-4s %3s A%4d %8.3f%8.3f%8.3f", \
"ATOM", ii, type[ii], res[ii], resID[ii], x[ii], y[ii], z[ii])
printf fmt, txt, dEcou[resLig[i]], dEvdw[resLig[i]] > Fout"~COU+VDW.pdb"
printf fmt, txt, PBres[resLig[i]], SAres[resLig[i]] > Fout"~PB+SA.pdb"
printf fmt, txt, dEcou[resLig[i]]+dEvdw[resLig[i]], \
PBres[resLig[i]]+SAres[resLig[i]] > Fout"~res_MM+PBSA.pdb"
printf fmt, txt, 0, dEcou[resLig[i]]+dEvdw[resLig[i]] \
+PBres[resLig[i]]+SAres[resLig[i]] > Fout"~res_MMPBSA.pdb"
}
}
}
function dimAPBS(file, Imol, minX, maxX, minY, maxY, minZ, maxZ) {
lenX=max(maxX-minX, 0.1); cntX=(maxX+minX)/2
lenY=max(maxY-minY, 0.1); cntY=(maxY+minY)/2
lenZ=max(maxZ-minZ, 0.1); cntZ=(maxZ+minZ)/2
cX =lenX*cfac; fX =min(cX, lenX+fadd)
cY =lenY*cfac; fY =min(cY, lenY+fadd)
cZ =lenZ*cfac; fZ =min(cZ, lenZ+fadd)
levN=4 # 划分级别
t=2^(levN+1)
nX=round(fX/df)-1; nX=max(t*round(nX/t)+1, 33)
nY=round(fY/df)-1; nY=max(t*round(nY/t)+1, 33)
nZ=round(fZ/df)-1; nZ=max(t*round(nZ/t)+1, 33)
if(gridType==0) { # GMXPBSA method
fpre=1; cfac=1.7
fX=lenX+2*fadd; cX=fX*cfac; nX=t*(int(fX/(t*df))+1+fpre)+1
fY=lenY+2*fadd; cY=fY*cfac; nY=t*(int(fY/(t*df))+1+fpre)+1
fZ=lenZ+2*fadd; cZ=fZ*cfac; nZ=t*(int(fZ/(t*df))+1+fpre)+1
}
MGset="mg-auto"
mem = 200*nX*nY*nZ/1024./1024. # MB
# npX=nX; npY=nY; npZ=nZ
# gmem=4000
# ofrac=0.1
# if(mem>=gmem) {
# while(mem>gmem) {
# maxN=max(npX, max(npY, npZ))
# if(maxN==npX) npX = t*((npX-1)/t-1)+1
# else if(maxN==npY) npY = t*((npY-1)/t-1)+1
# else if(maxN==npZ) npZ = t*((npZ-1)/t-1)+1
# mem = 200*npX*npY*npZ/1024./1024
# }
# t=nX/npX; if(t>1) npX = int(t*(1+2*ofrac) + 1.0);
# t=nY/npY; if(t>1) npY = int(t*(1+2*ofrac) + 1.0);
# t=nZ/npZ; if(t>1) npZ = int(t*(1+2*ofrac) + 1.0);
# MGset="mg-para\n ofrac "ofrac"\n pdime "npX" "npY" "npZ
# }
XYZset=" "MGset \
"\n mol "Imol \
"\n dime "nX" "nY" "nZ" # 格点数目, 所需内存: "mem" MB" \
"\n cglen "cX" "cY" "cZ" # 粗略格点长度" \
"\n fglen "fX" "fY" "fZ" # 细密格点长度" \
"\n fgcent "cntX" "cntY" "cntZ" # 细密格点中心" \
"\n cgcent "cntX" "cntY" "cntZ" # 粗略格点中心"
return \
"ELEC name "file"\n" \
XYZset "\n" \
PBEset "\n" \
"end\n\n" \
"ELEC name "file"~VAC\n" \
XYZset "\n" \
PBEset0 "\n" \
"end\n\n" \
"APOLAR name "file"~SAS\n" \
" mol "Imol"\n" \
PBAset"\n" \
"end\n\n" \
"print elecEnergy "file" - "file"~VAC end\n" \
"print apolEnergy "file"~SAS end\n\n"
}
function min(x, y) { return xy ? x : y }
function round(x) { return int(x+0.5) }
' _$pid.pdb
fi
################################################################################
# 4. 删除临时文件
# 4. remove intermediate files
################################################################################
#rm -f $trjwho.xtc $trjcnt.xtc $trjcls.xtc
#rm -f io.mc _$pid.pdb $scr $qrv \#_$pid*\#
3.7 MtD
1.Metadynamics(MtD)可以使用PLUMED 和Colvars等分子模拟计算自由能的插件,限于篇幅问题下次更新做单独的更新
3.8.进阶教程:使用gromacs计算配体与受体的结合自由能
1.设置
该过程如下图所示,在文章1中已经进行了详细的描述,可前去了解,这里只挂出示意图:

在这个热力学循环中,我们需要模拟的系统由它们周围的黑色圆圈表示,限制势的存在由回形针指示,白色配体意味着它不与环境相互作用(但仍存在分子内作用),蓝色表示显示水溶剂环境。
A 状 态 : L + ( W a t e r ) − − − > L e l e c + v d w A状态:L+(Water)--->L_{elec+vdw} A状态:L+(Water)−−−>Lelec+vdw
A状态下:假设蛋白质(P)与小分子(L)远离,此时L与P之间还没有相互作用,L体系与环境(如溶剂水)间具有相互作用(橙色表示有相互作用:L与水耦合)主要为静电相互作用L(elec),非静电相互作用(VDW)为溶剂化自由能(ΔG(solve))
B 状 态 : L e l e c + v d w − − − − − > L + ( W a t e r ) B状态:L_{elec+vdw}----->L+(Water) B状态:Lelec+vdw−−−−−>L+(Water)
B状态:分别慢慢关闭 L与环境(Water)间的相互作用,即小分子 -溶剂间的 ele和 vdw,此时的小分子如同虚原子(Ghost-atoms,白色表示小分子此时与环境无相互作用:L与水解耦,即为去溶剂化);
C 状 态 : L − − − − − − > L ( r e s t r ) C状态:L------>L_(restr) C状态:L−−−−−−>L(restr)
C状态 :无相互作用力的小分子与蛋白结合,进入蛋白的 结合口袋,形成状态D中的复合物,这里引入了限制势,限制势的引入是为了防止配体在蛋白质中乱跑(尤其是关闭了大部分配体与环境的相互作用时)、阻碍收敛,只要最后在能量加上校正项就行。
需要注意的是在关闭相互作用时,我们总是先关闭静电作用,再关闭非静电项;如果反过来的话,那就会出现这样的情况——正负电荷的原子可能会靠的无限近,这显然是不合理的。反之,如果使用的是逐步打开相互作用的途径,那就应该先打开非静电项,再打开静电项。
D 状 态 : P + L r e s t r − − − − − − > P L ( r e s t r ) D状态:P+L_{restr}------>PL(restr) D状态:P+Lrestr−−−−−−>PL(restr)
C状态 :看上去我们似乎C到D的自由能变化,但实际上C与D的状态是完全相同的,反正此时的配体都不与周围环境有任何相互作用了,由自由能的定义可知,换个环境对自由能改变为0。
E 状 态 : P L − − − − − − − > P L r e s t r + e l e c + v d w E状态:PL------->PL_{restr+elec+vdw} E状态:PL−−−−−−−>PLrestr+elec+vdw
**E状态 :**L在P的结合口袋中,打开配体L与受体P之间的相互作用elec和VDW,此时ΔG为结合自由能ΔG(bind)
F 状 态 : P L r e s t r + e l e c + v d w − − − − − − > P L e l e c + v d w F状态:PL_{restr+elec+vdw}------>PL_{elec+vdw} F状态:PLrestr+elec+vdw−−−−−−>PLelec+vdw
**状态F:**解除限制势L与P间的限制势,形成闭合的热力学循环,形成生物体系中的配受体复合物
因此,由 A 直接到F变化的结合自由能 Δ G ( b i n d i n g ) \Delta G_(binding) ΔG(binding),可等价于由状 态 A 经状态 B—E(C->D过程自由能无变化)转变为状态F过程的能量变化总和。(公式2.26所示)
Δ G b i n d i n g 0 = G F − G A (3.8.1) \Delta G_{binding}^0 =G_F-G_A \tag {3.8.1} ΔGbinding0=GF−GA(3.8.1)
Δ G e l e c + v d w s o l v = G B − G A (3.8.2) \Delta G_{elec+vdw}^{solv}=G_B-G_A \tag {3.8.2} ΔGelec+vdwsolv=GB−GA(3.8.2)
Δ G r e s t r s o l v = G c − G B (3.8.3) \Delta G_{restr}^{solv}=G_c-G_B \tag {3.8.3} ΔGrestrsolv=Gc−GB(3.8.3)
Δ G = 0 = G D − G C (3.8.4) \Delta G=0=G_D-G_C \tag {3.8.4} ΔG=0=GD−GC(3.8.4)
Δ G e l e c + v d w p r o t = G E − G D (3.8.6) \Delta G_{elec+vdw}^{prot}=G_E-G_D \tag {3.8.6} ΔGelec+vdwprot=GE−GD(3.8.6)
Δ G r e s t r s o l v = G F − G E (3.8.7) \Delta G_{restr}^{solv}=G_F-G_E \tag {3.8.7} ΔGrestrsolv=GF−GE(3.8.7)
以上(3.8.2)到(3.8.3)式相加得:
Δ G e l e c + v d w s o l v + Δ G r e s t r s o l v + Δ G + Δ G e l e c + v d w p r o t + Δ G r e s t r s o l v = G F − G A = Δ G b i n d i n g 0 (3.8.8) \Delta G_{elec+vdw}^{solv}+\Delta G_{restr}^{solv}+\Delta G+\Delta G_{elec+vdw}^{prot}+\Delta G_{restr}^{solv}=G_F-G_A=\Delta G_{binding}^0 \tag {3.8.8} ΔGelec+vdwsolv+ΔGrestrsolv+ΔG+ΔGelec+vdwprot+ΔGrestrsolv=GF−GA=ΔGbinding0(3.8.8)
通过分别模拟小分子在溶剂水和靶蛋白的口袋 中,慢慢关闭或打开小分子与环境间的相互作用 ( ele和 vdw),以上两个过程构成了一个热力学循 环。由于小分子与环境间相互作用的关闭(或打开) 的过程是不存在的非真实物理过程,因此该计算过程也被称为“炼金术”。(使用不同的的算法来评估这一过程的变化,从而会衍生出不同的自由能计算方法,在第3节中讲到)
由上图所示,依据分子动力学模拟计算自由能的原理知,我们无需模拟配体从真空环境中到水溶剂环境中(溶剂化),然后在去溶剂化,与配体结合这一系列的过程,我们只需要取其中几个终状态,然后人为的去开关体系中的作用项即可模拟出这些过程中的变化,在本教程中主要涉及以下三个:
1.配体与溶液的解耦:A---->B状态
Δ G 1 = Δ G e l e c + v d w s o l v = G B − G A (3.8.9) \Delta G_1=\Delta G_{elec+vdw}^{solv}=G_B-G_A \tag{3.8.9} ΔG1=ΔGelec+vdwsolv=GB−GA(3.8.9)
2.约束:B---->C状态
在解耦配体(状态B到C)上加上约束,可以用论文中的32式来分析考虑(当使用这组特定的约束时)。
Δ G 2 = Δ r e s t r o n s o l v e = R T ln [ 8 π 2 V 0 r 0 2 s i n θ A , 0 , s i n θ B , 0 , ( K r K θ A K θ B K ϕ A K ϕ B K ϕ C ) 1 2 ( 2 π k T ) 3 ] (3.8.10) \Delta G_2=\Delta_{restr_{on}}^{solve}=RT\ln [\frac{8π^2V^0}{r_{0}^{2}sin \theta_{A,0},sin \theta_{B,0},} \frac{(K_rK_{\theta A} K_{\theta B}K_{\phi A}K_{\phi B}K_{\phi C})^{\frac{1}{2}}} {(2πkT)^3}] \tag {3.8.10} ΔG2=Δrestronsolve=RTln[r02sinθA,0,sinθB,0,8π2V0(2πkT)3(KrKθAKθBKϕAKϕBKϕC)21](3.8.10)
R R R 为理想气体常数
K K K为以开尔文为单位的温度。
V 0 V^{0} V0为对应于一摩尔标准态(1660 A ˚ 3 Å^{3} A˚3)的体积。
r 0 r_0 r0为是约束装置的参考距离。
θ A , θ B \theta_A,\theta_B θA,θB为是约束的参考角度。
k x k_x kx为是距离( r 0 r_0 r0)的力常数,我们应用两个角度( θ A , θ B \theta_A,\theta_B θA,θB)和三个二面角( ϕ A , ϕ B , ϕ C \phi A,\phi B,\phi C ϕA,ϕB,ϕC)的限制
3.配体与受体解耦:
正常的模拟过程应该是配体与受体耦合,但是为了处理简单使用的是配体与受体已经结合在一起的模型,所以这里模拟是解耦的过程(F—>D)状态,与配体与受体结合过程(D—>F)相反,所以该过程的自由能要取反:
Δ G 3 = − Δ G e l e c + v d w + r e s t r p r o t (3.8.11) \Delta G_3=-\Delta G_{elec+vdw+restr}^{prot} \tag {3.8.11} ΔG3=−ΔGelec+vdw+restrprot(3.8.11)
设置:
1.既然需要进行两个独立体系自由能计算,最后通过热力学循环来求得配体与受体之间绝对结合自由能,那么就需要去准备两套模拟配置文件:如下:
1.对于配体与溶液的解耦:A---->B状态
**01.ligand文件下的目录结构**
├── collect_dhdl.sh :批量收集分析所需要数据的脚本
├── batche.sh :批量执行job_0.sh -->job_19.sh 的脚本
├── write_mdp.sh :批量生成mdp文件的脚本,主要修改mdp 中的init_lambda_state =nlambda 的值
├── write_sh.sh :批量生成job文件的脚本,主要修改job文件中 LAMBDA=Wnum的值,
├── job.sh :为每一窗口独立运行整个模拟过程(EM->NVT->NPT->Production_MD),即Lambda_0,
├── MDP :总运行参数mdp文件夹
│ ├── EM :能量最小化参数mdp文件夹
│ │ ├── enmin.mdp :第1次能量最小化参数mdp文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh enmin
│ ├── NVT :等温等容预平衡参数mdp文件夹
│ │ ├── nvt.mdp :等温等容预平衡参数mdp文件
│ │ ├── write_mdp.sh : 用法:./write_mdp.sh nvt 即可后接mdp文件前缀
│ ├── NPT :等温等压预平衡参数mdp文件夹
│ │ ├── npt.mdp :等温等压预平衡参数mdp文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh npt 即可后接mdp文件前缀
│ └── Production_MD :成品模拟参数mdp文件夹
│ ├── prod.mdp :成品模拟参数mdp文件
│ └── write_mdp.sh : 用法:./write_mdp.sh prod 即可后接mdp文件前缀
├── Ligand :结构文件和拓扑文件夹
│ ├── ligand.gro :含1个甲烷水盒子结构文件
│ ├── posre_ligand.itp :约束文件
│ └── ligand.top :体系top文件
└── REDERME.md
以为该体系enmin.mdp为例子,说明配体与溶液发生去耦合过程的自由能计算代码设置,每项具体信息见3.3
在.mdp文件中,我们定义了2个lambda向量:coulomb和vdw
;====================================================
; Energy minimization
;====================================================
;----------------------------------------------------
; RUN CONTROL & MINIMIZATION
;----------------------------------------------------
define = -DFLEXIBLE
integrator = steep
nsteps = 5000
emtol = 100
emstep = 0.01
nstcomm = 100
;----------------------------------------------------
; OUTPUT CONTROL
;----------------------------------------------------
nstxout = 250 ; save coordinates to .trr every 250 steps
nstvout = 0 ; don't save velocities to .trr
nstfout = 0 ; don't save forces to .trr
nstxout-compressed = 500 ; xtc compressed trajectory output every 500 steps
compressed-x-precision = 1000
nstlog = 500 ; update log file every 500 steps
nstenergy = 500 ; save energies every 500 steps
nstcalcenergy = 100
;----------------------------------------------------
; NEIGHBOR SEARCHING
;----------------------------------------------------
cutoff-scheme = Verlet
ns-type = grid
nstlist = 1
rlist = 1.0
;----------------------------------------------------
; BONDS
;----------------------------------------------------
constraints = none
;----------------------------------------------------
; ELECTROSTATICS
;----------------------------------------------------
coulombtype = PME
rcoulomb = 1.0
pme-order = 6
fourierspacing = 0.10
ewald-rtol = 1e-6
;----------------------------------------------------
; VDW
;----------------------------------------------------
vdw-type = PME
rvdw = 1.0
vdw-modifier = Potential-Shift
ewald-rtol-lj = 1e-3
lj-pme-comb-rule = Geometric
DispCorr = EnerPres
;----------------------------------------------------
; TEMPERATURE & PRESSURE COUPL
;----------------------------------------------------
Tcoupl = no
Pcoupl = no
gen_vel = no
;----------------------------------------------------
; FREE ENERGY CALCULATIONS
;----------------------------------------------------
free-energy = yes
couple-moltype = ligand
couple-lambda0 = vdw-q ;当lambda=0(A状态),范德华和静电相互作用打开
couple-lambda1 = none ;当lambda=1(B状态),所有作用力关闭
; A---->B,配体与受体解耦
couple-intramol = no
separate-dhdl-file = yes
sc-alpha = 0.5
sc-power = 1
sc-sigma = 0.3
init-lambda-state = nlambda ;nlambda的值从0-19
coul-lambdas = 0.0 0.25 0.5 0.75 1.0 1.00 1.0 1.0 1.0 1.0 1.0 1.0 1.00 1.0 1.00 1.0 1.00 1.0 1.00 1.0
vdw-lambdas = 0.0 0.00 0.0 0.00 0.0 0.05 0.1 0.2 0.3 0.4 0.5 0.6 0.65 0.7 0.75 0.8 0.85 0.9 0.95 1.0
nstdhdl = 100
calc-lambda-neighbors = -1
3.对于配体与受体解耦:
**02.Complex文件下的目录结构**
├── collect_dhdl.sh :批量收集分析所需要数据的脚本
├── batche.sh :批量执行job_0.sh -->job_19.sh 的脚本
├── write_mdp.sh :批量生成mdp文件的脚本,主要修改mdp 中的init_lambda_state =nlambda 的值
├── write_sh.sh :批量生成job文件的脚本,主要修改job文件中 LAMBDA=Wnum的值,
├── job.sh :为每一窗口独立运行整个模拟过程(EM->NVT->NPT->Production_MD),即Lambda_0,
├── MDP :总运行参数mdp文件夹
│ ├── EM :能量最小化参数mdp文件夹
│ │ ├── enmin.mdp :第1次能量最小化参数mdp文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh enmin
│ ├── NVT :等温等容预平衡参数mdp文件夹
│ │ ├── nvt.mdp :等温等容预平衡参数mdp文件
│ │ ├── write_mdp.sh : 用法:./write_mdp.sh nvt 即可后接mdp文件前缀
│ ├── NPT :等温等压预平衡参数mdp文件夹
│ │ ├── npt.mdp :等温等压预平衡参数mdp文件
│ │ └── write_mdp.sh : 用法:./write_mdp.sh npt 即可后接mdp文件前缀
│ └── Production_MD :成品模拟参数mdp文件夹
│ ├── prod.mdp :成品模拟参数mdp文件
│ └── write_mdp.sh : 用法:./write_mdp.sh prod 即可后接mdp文件前缀
├── Complex :结构文件和拓扑文件夹
│ ├── complex.gro :含1个甲烷水盒子结构文件
│ ├── ligand.itp :配体拓扑文件
│ ├── posre_ligand.itp :约束文件
│ ├── posre_complex.itp :约束文件
│ └── complex.top :体系top文件
└── REDERME.md
同样以为该体系enmin.mdp为例子,说明配体与溶液发生去耦合过程的自由能计算代码设置,每项具体信息见3.3
在.mdp文件中,我们定义了3个lambda向量:bonded,coulomb和vdw
;====================================================
; Energy minimization
;====================================================
;----------------------------------------------------
; RUN CONTROL & MINIMIZATION
;----------------------------------------------------
define = -DFLEXIBLE
integrator = steep
nsteps = 5000
emtol = 100
emstep = 0.01
nstcomm = 100
;----------------------------------------------------
; OUTPUT CONTROL
;----------------------------------------------------
nstxout = 250 ; save coordinates to .trr every 250 steps
nstvout = 0 ; don't save velocities to .trr
nstfout = 0 ; don't save forces to .trr
nstxout-compressed = 500 ; xtc compressed trajectory output every 500 steps
compressed-x-precision = 1000
nstlog = 500 ; update log file every 500 steps
nstenergy = 500 ; save energies every 500 steps
nstcalcenergy = 100
;----------------------------------------------------
; NEIGHBOR SEARCHING
;----------------------------------------------------
cutoff-scheme = Verlet
ns-type = grid
nstlist = 1
rlist = 1.0
;----------------------------------------------------
; BONDS
;----------------------------------------------------
constraints = none
;----------------------------------------------------
; ELECTROSTATICS
;----------------------------------------------------
coulombtype = PME
rcoulomb = 1.0
pme-order = 6
fourierspacing = 0.10
ewald-rtol = 1e-6
;----------------------------------------------------
; VDW
;----------------------------------------------------
vdw-type = PME
rvdw = 1.0
vdw-modifier = Potential-Shift
ewald-rtol-lj = 1e-3
lj-pme-comb-rule = Geometric
DispCorr = EnerPres
;----------------------------------------------------
; TEMPERATURE & PRESSURE COUPL
;----------------------------------------------------
Tcoupl = no
Pcoupl = no
gen_vel = no
;----------------------------------------------------
; FREE ENERGY CALCULATIONS
;----------------------------------------------------
free-energy = yes
couple-moltype = ligand
couple-lambda0 = vdw-q
couple-lambda1 = none
couple-intramol = no
separate-dhdl-file = yes
sc-alpha = 0.5
sc-power = 1
sc-sigma = 0.3
init-lambda-state = nlambda ; ;nlambda的值从0-29
bonded-lambdas = 0.0 0.01 0.025 0.05 0.075 0.1 0.2 0.35 0.5 0.75 1.0 1.00 1.0 1.00 1.0 1.00 1.0 1.0 1.0 1.0 1.0 1.0 1.00 1.0 1.00 1.0 1.00 1.0 1.00 1.0
coul-lambdas = 0.0 0.00 0.000 0.00 0.000 0.0 0.0 0.00 0.0 0.00 0.0 0.25 0.5 0.75 1.0 1.00 1.0 1.0 1.0 1.0 1.0 1.0 1.00 1.0 1.00 1.0 1.00 1.0 1.00 1.0
vdw-lambdas = 0.0 0.00 0.000 0.00 0.000 0.0 0.0 0.00 0.0 0.00 0.0 0.00 0.0 0.00 0.0 0.05 0.1 0.2 0.3 0.4 0.5 0.6 0.65 0.7 0.75 0.8 0.85 0.9 0.95 1.0
nstdhdl = 100
calc-lambda-neighbors = -1
模拟
模拟过程参考3.3.,这里不在赘述
分析
分析过程参考3.3.,可以使用alchemical_analysis快捷分析。
根据公式3.8.8对配体与受体之间的结合自由能定义可以知:
Δ G b i n d i n g 0 = Δ G 1 + Δ G 2 + Δ G 3 = Δ G e l e c + v d w s o l v + Δ r e s t r o n s o l v e − Δ G e l e c + v d w + r e s t r p r o t \begin{aligned} \Delta G_{binding}^0&=\Delta G_1+\Delta G_2+\Delta G_3 \\ &=\Delta G_{elec+vdw}^{solv}+\Delta_{restr_{on}}^{solve}-\Delta G_{elec+vdw+restr}^{prot} \end{aligned} ΔGbinding0=ΔG1+ΔG2+ΔG3=ΔGelec+vdwsolv+Δrestronsolve−ΔGelec+vdw+restrprot
其中 Δ G 1 和 Δ G 3 \Delta G_1和\Delta G3 ΔG1和ΔG3的值可以通过本例分子模计算自由能再使用alchemical_analysis工具分析得出,这里不赘述,同3.3, Δ G 2 \Delta G_2 ΔG2的值可以通过公式3.8.10得出,具体见原教程1.gromacs计算结合自由能(en)
备注:
gromacs版本不同,本教程所以案例使用的是gmx 版本,请根据实际情况做修改,常见版本如下:
gmx : MD引擎二进制,没有MPI,但支持线程-MPI。如果GROMACS在一个节点上执行,以及在计算节点上运行分析工具,则非常有用。
gmx_mpi : 支持MPI的MD引擎二进制。这是研究人员最常使用的二进制。
gmx_d : 没有MPI的MD引擎双精度二进制,但支持线程-MPI。
gmx_mpi_d : 和上面一样,但是是双精度的。这个比混合精度的mdrun_mpi慢很多。
gmx_avx : 这个二进制文件在不支持AVX2的情况下产生,用于登录节点。
参考链接:
1.gromacs手册:7.3.23 自由能计算
2.gromacs手册:自由能计算的实现
3.gromacs计算结合自由能案例(en)
4.gmx bar: 利用Bennett接受比率方法计算自由能差的估计值(翻译: 陈珂)
以上所有示例模拟文件配置文件 关注:“理论与计算化学初学者”公众号,回复“自由能”获得下载链接