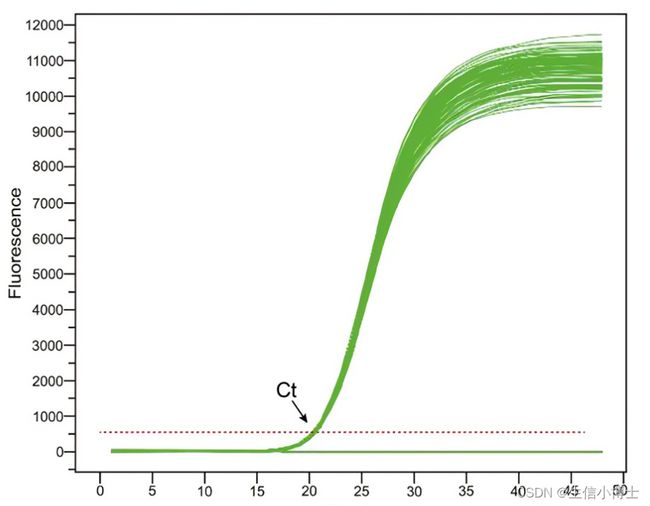
qPCR(荧光定量PCR)的Ct值
今天我们要说的问题,也是对qPCR的更高阶的认识。
1、Ct值到底是不是YYDS?
2、Ct值跟哪些因素有关?
3、同样的模板,Ct值大试剂盒就差吗?要不要换试剂盒?

还是从这个盗版来的图说起。你必须明白以下几个概念和问题。
Ct值-阈值-基线
1、阈值循环数(Cycle threshold,Ct值)的含义为:每个反应管内的荧光信号到达设定阈值时所经历的循环数。Ct值与模板的起始拷贝数的对数存在线性关系,起始模板浓度越高,Ct值越小;起始模板量浓度越低,Ct值越大。这就是荧光定量PCR的理论基础。
2、阈值怎么确定呢?阈值(threshold)一般是基线的标准偏差的10倍。为什么是10倍,而不是5倍8倍?就是为了和基线拉开距离,证明PCR扩增进入指数扩增期。为什么不是20倍30倍?因为每个一PCR循环都会放大误差,而此时误差还未被放大。我们当然可以把阈值设置到30个循环,但是30个循环以后,扩增效率和其它误差带来的数据偏差,估计他妈妈都不认识了。
3、基线又是怎么确定呢?基线(baseline)通常是3-15个循环的荧光信号,同一次反应中针对不同的基因需单独设置基线。
很显然,基线一变,阈值就改变了,阈值改变,Ct值就跟着改变。由于每次实验的样本和仪器,甚至混合情况不一样,使得基线有所差异,最终会影响Ct值。所以拿上一次实验的样本去检测本次实验的Ct值是否一致,本身就是刻舟求剑,没有可比性,所以童鞋们应该明白了,为啥qPCR一做就要放在一起做,就是这个原因。
Ct值跟哪些因素有关?
按照影响的严重性进行描述:仪器<耗材<试剂<<<样本浓度,越往后面越严重。这是肯定的了,如果仪器耗材试剂都能很大程度上影响对样本浓度的测定,那这就是一个失败的测试。
仪器:很显然,仪器对荧光信号检测的敏感度,以及计算软件,都会影响到Ct值。我们一旦买了一台仪器,这些因素就已经确定了。高校实验室的仪器几乎都是清一色的进口品牌:ABI、Bio-Rad、Roche,几十万一台,你几乎无法找他们的麻烦。
耗材:很多童鞋会忽略耗材的影响,其实耗材的选择是非常重要的,荧光信号强不强,先过耗材这一关,你用的板子是什么板?白色or透明;用的好是数据看板,用的不好就是仙人板板。用的膜是什么膜?高透or普通;好的膜就跟没有一样,就如同管理,最好的管理就是没有管理”太上,不知有之“。适配性怎么样?我们曾经做过一个qPCR金标准耗材的选择表,感兴趣的可以找我(Thankyou_baby)要一份。
试剂:这是大家最容易被欺骗的地方,认为试剂好Ct值就比较低,因为只有这个指标是最直观的,以至于市面上的公司都在拼命的降低Ct值,有的公司甚至以增加预热,预反应的方式,让Taq酶启动起来,先反应个几个循环。
在此,我跟大家讲讲试剂的内在本质。
1.Taq酶:一般来说,做qPCR所使用的Taq酶都是热启动的Taq酶,热启动的Taq酶分为两种,一种是化学修饰,一种是抗体修饰,很显然,抗体修饰的热启动酶更高级,生物学活动要比化学活动敏感得多,一旦到达反应温度,抗体修饰的热启动酶即可释放活性,而化学修饰有短暂的反应时间。所以,只要一个荧光定量PCR试剂盒的酶是抗体修饰的,那么大概率Ct值比化学修饰的要低。

两种热启动Taq示意图
化学修饰是上一个世纪的技术。但是化学修饰并不是一无是处,它能够保证酶活在反应过程中得到补充,特别是在最后的十几个循环。这就是为什么有的公司会添加两种酶。从某种意义上讲,它也是对抗体修饰酶活的不自信。如果一个酶能够持续反应到60个循环,添加候补酶就大可不必,就像一个足球运动员,如果它能够精神饱满的跑完90分钟,替补人员就没必要使用了。另外,如果抗体修饰的酶性能卓著,添加其他的酶反而是占用酶浓度。生产商肯定考虑过这个问题。
2.镁离子浓度:镁离子浓度是Taq酶活性的调节剂,合适的镁离子浓度能够促进Taq酶活性释放,浓度过低,会显著降低酶活性;浓度过高又使酶催化非特异性扩增增强。镁离子浓度还会影响引物的退火、模板与PCR产物的解链温度,从而影响扩增片段的产率。镁离子离子的浓度一般控制在25mM,当然了,一个好的试剂盒,镁离子浓度一定是控制的比较好的。有的商家在试剂里面加入镁离子的螯合剂,可以达到镁离子浓度自动调节的作用。
3.Buffer:你看到的市面上大多数厂家的试剂盒的酶,可能都是同一厂家批量生产,然而其性能更卓越,主要是优化了Buffer,具体如何优化,我也不知道,反正你拿到产品也无法优化。
4.dNTP:主要考虑dNTP的纯度。这方面不得不说某些国外厂家专门生产dNTP,卖给中国的公司包装成试剂盒,你可以问问生产商dNTP的来源。纯度不够,抑制PCR反应。
试剂对Ct值影响主要是这几个方面。作为试剂的消费者,其实要考虑的不是Ct值,而是该试剂盒在低浓度模板和高浓度模板的情况下,是否会产生PCR抑制。从而使得标准曲线之间的距离不均衡。其实有个公司的产品是业界良心,一般生产商用中国制造贴美国品牌,而这个生产商用国外的高级原料包装成国产试剂,卖国产的价格,反向补贴,真是业界良心,为了避免种草,此处不公开说明。
样本浓度
首先我们说一般性原理:PCR通过Taq酶进行DNA合成,经过一个反应,扩增片段DNA的浓度翻一倍,也就是说每扩增N次,产物浓度就会变成2的N次方。
- 产物量Cn=起始模板量C1×2^循环数n
然而并不是每次都能100%的翻倍扩增,要不然为什么前面15个循环都在基线期呢?细思极恐。在扩增过程中造成的损失,我们用扩增效率(E)来表示,把2拆分成(1+E),通常,我们认为扩增效率与Taq酶有关,这其实是一个笼统的看法。我们都知道一个反应体系包含Taq酶,镁离子,buffer,dNTP,水,另外就是引物和模板,任何一个元素都可能影响扩增效率。
- 产物量Cn=起始模板量C1×(1+E)^循环数n
扩增效率E到底能有多少呢?如果是100%,那就是1,再好不过了,大多数能够做到95%~100%,这已经是很不错的扩增效率了。但是莫名其妙的是有的实验做到了110%,你可以用小脑袋瓜思考一下,DNA的半保留复制,一个复制循环竟然复制出多余2倍的DNA,怪了。很明显,引物二聚体可能在复制,非特异性扩增DNA可能也在复制,因为我们检测的是荧光值,只有多于目的片段的DNA在复制,才能可突破100%。
做一个已知样本量的标准曲线,倍比稀释模板,就会得到如下的结果。

模拟数据
- 标准曲线制作:利用Mean Ct (平均值)作图可得到标准曲线
- y = -3.29x + 40.33;纵坐标y就是Ct值,横坐标x就是样本浓度的数量级,3.29是斜率Slope,当扩增效率为100%时斜率为-3.32。
- R2 = 0.9978;相关系数Correlation coefficient (R^2) 表明标准曲线的线性,通常要求>0.98,越接近1,结果可信度越高。
- 斜率-�=10−1/斜率-1 ;越接近1越理想。
你也许会问,这些数据依据是如何得来的,为什么是这个公式,答:高中数学。咱们做生物学研究的,数学一般都不咋地,老一辈科学家都已经做出来了,你跟着公式计算没问题。

标准曲线
所以,回到我们的问题,除了最重要的样本浓度,仪器、耗材、试剂都会对Ct值造成影响。
Ct值对我们计算样本的拷贝数影响到底有多大呢?以下是在扩增效率完美达到100%的情况下和真实情况下的比较:
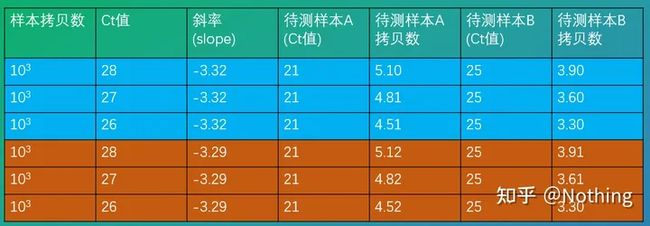
数字模拟
- 同样的扩增效率,Ct值少一个循环,拷贝数的数量级差别大概是0.3,也就是2倍左右;
- 同样的Ct值,扩增效率只要不是特别拉跨,拷贝数的数量级差别是0.01,也就是1倍左右;
- 当然这是理论计算,真实情况下扩增效率和Ct值相互影响;
结论:选择Ct值较低的产品是正确的,但是一定要避免非特异性扩增或者引物二聚体的情况。
解决方案
- 如果扩增效率比较高,Ct值降低是自然而然的事情;
- 如果遇到扩增效率大于1,要注意检查引物,它会不会产生二聚体,它的特异性如何,是不是要重新设计,因为扩增效率是根本;
- 扩增效率不高的主要原因有:热启动Taq酶活性不足、dNTP不纯、没有调校好Buffer、以及模板不纯,存在PCR扩增抑制,建议更换试剂盒