RNA-seq分析服务器安装生信工具过程
一.测试服务器设定
1.使登录后自动进入/home目录下
vim ~/.bashrc
#在文件中加入以下行后保存退出
cd /home2.新建RNAseq_tool文件夹,存放各工具
mkdir RNAseq_tool二.各生信工具在测试服务器下的安装
1.sratoolkit下载及安装
#下载并解压
wget "ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz"
tar -xzf sratoolkit.current-centos_linux64.tar.gz
#进入bin目录
cd [download_location]/sratoolkit[version]/bin/
#设置path并使其生效
vi /etc/profile
增加以下行
export PATH=$PATH:/home/RNAseq_tool/sratoolkit.2.8.2-ubuntu64/bin
vi /etc/environment
增加以下行:
PATH="/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin:/bin:/usr/games:/usr/local/games:/home/RNAseq_tool/sratoolkit.2.8.2-1-ubuntu64/bin"
source /etc/profile
source /etc/enviroment
#测试安装是否成功
fastq-dump –version
#运行案例
fastq-dump -X 5 -Z SRR390728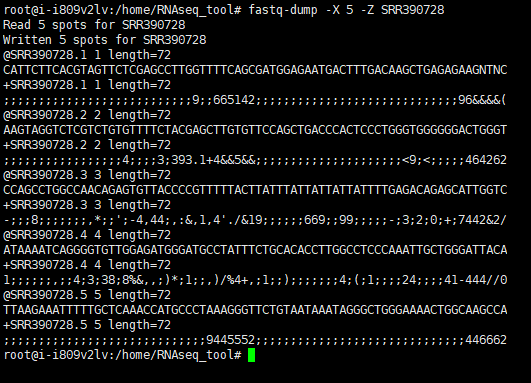
#配置SRAtoolkit的下载路径
[安装路径]/bin/vdb-config –i 设定下载路径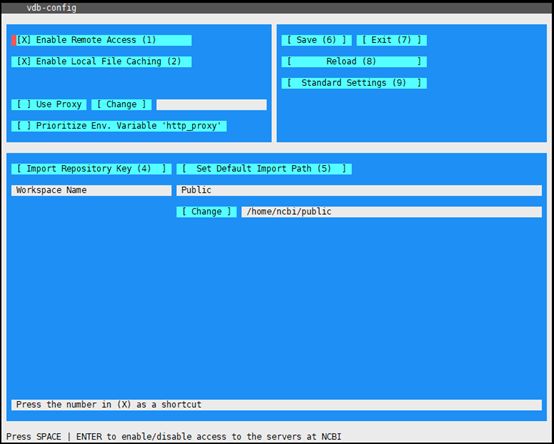
2.fastqc下载及安装
#fastqc需要java环境,首先下载并配置java,此处下载 jre1.8.0_144
wget http://javadl.oracle.com/webapps/download/AutoDL?BundleId=225345_090f390dda5b47b9b721c7dfaa008135
#进入安装目录解压
tar –xzvf jre-8u144-linux-x64.tar.gz
#配置java的环境变量
vi /etc/profile
添加以下行
JAVA_HOME=/usr/java/jre1.8.0_144/
export CLASSPATH=.:$JAVA_HOME/lib:$CLASSPATH
export PATH = $PATH:$JAVA_HOME/bin
vi /etc/environment
添加java安装目录,此时文件内容为:
PATH="/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin:/bin:/usr/games:/usr/local/games:/home/RNAseq_tool/sratoolkit.2.8.2-1-ubuntu64/bin:/usr/java/jre1.8.0_144/bin"
#下载及解压
wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip
unzip fastqc_v0.11.5.zip
cd FastQC
#添加执行权限
chmod 755 fastqc
#建立软连接
ln -s [download_location]/FastQC/fastqc /usr/local/bin/fastqc
#测试fastqc是否正常运行
fastqc SRR390728.fastq #运行结果如下图所示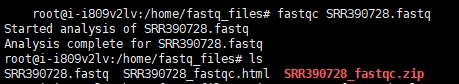
处理好的SRR390728_fastqc.html可以通过WinSCP下载到Windows系统上进行查看。
3.cutadapt下载及安装
#安装python-pip,python-dev
apt-get install python-pip python-dev
#使用pip安装cutadapt,该方法将cutadapt二进制文件安装到./usr/.local/bin中,无需设置环境变量
pip install --user --upgrade cutadapt
#尝试运行
cutadapt --version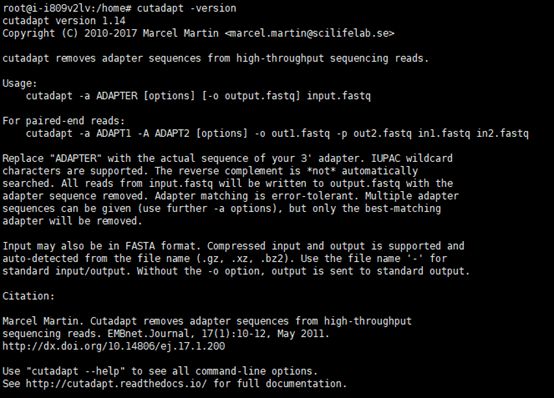
4.fastx-toolkit下载及安装
#下载并安装依赖库libgtextutils 0.7
wget https://github.com/agordon/libgtextutils/releases/download/0.7/libgtextutils-0.7.tar.gz
tar –xzvf libgtextutils-0.7.tar.gz
#进入解压后文件夹进行编译安装
./configure
make
make install
#下载并解压fastx 0.0.14:
wget https://github.com/agordon/fastx_toolkit/releases/download/0.0.14/fastx_toolkit-0.0.14.tar.bz2
tar –xjvf fastx_toolkit-0.0.14.tar.bz2
#进入解压后文件夹进行编译安装
./configure
make
make install
#配置环境变量
vi /etc/profile
添加如下行
export LD_LIBRARY_PATH=/usr/local/lib:$LD_LIBRARY_PATH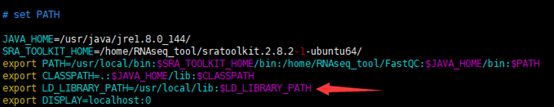
5.安装bowtie2(旧流程)
#安装依赖包libtbb2
apt-get install libtbb2
#下载并解压bowtie2
wget https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.3.2/bowtie2-2.3.2-linux-x86_64.zip/download
unzip bowtie2-2.3.2-linux-x86_64.zip
#配置环境变量
vi /etc/profile
export PATH=$PATH://home/RNAseq_tool/bowtie2-2.3.26.安装Tophat2(旧流程)
#下载并解压
wget http://ccb.jhu.edu/software/tophat/downloads/tophat-2.1.1.Linux_x86_64.tar.gz
tar –xzvf tophat-2.1.1.Linux_x86_64.tar.gz
#添加软连接
cd /usr/local/bin
ln –s /home/RNAseq_tool/tophat-2.1.1.Linux_x86_64/tophat2
#测试tophat2
#下载并解压测试数据
wget http://ccb.jhu.edu/software/tophat/downloads/test_data.tar.gz
tar –xzvf test_data.tar.gz
#测试:(实际使用时需要额外生成index文件)
cd test_data
tophat2 -r 20 test_ref reads_1.fq reads_2.fq7.安装HISAT2(Tophat替代工具)
#下载源码并解压
wget http://ccb.jhu.edu/software/hisat2/downloads/hisat2-2.0.0-beta-source.zip
unzip hisat2-2.0.0-beta-source.zip
#进入解压后目录并编译安装
cd hisat2-2.0.0-beta/
make
#添加环境变量并使其立即生效
export PATH=$PATH:/home/RNAseq_tool/hisat2-2.0.0-beta
source ~/.bashrc
#试运行
hisat2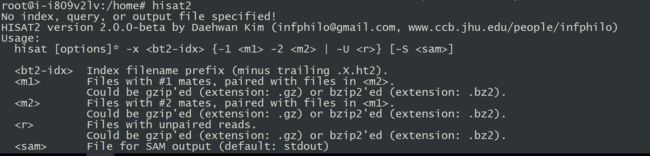
8.安装samtools
#下载依赖库:libncurses5-dev, zlib1g-dev libbz2-dev liblzma-dev
apt-get install libncurses5-dev zlib1g-dev libbz2-dev liblzma-dev
#下载samtools 1.5,bcftools 1.5,htslib 1.5:
wget https://github.com/samtools/samtools/releases/download/1.5/samtools-1.5.tar.bz2
wget https://github.com/samtools/bcftools/releases/download/1.5/bcftools-1.5.tar.bz2
wget https://github.com/samtools/htslib/releases/download/1.5/htslib-1.5.tar.bz2
#安装三个工具(分别进入各工具文件夹,进行编译安装,顺序:htslib>bcftools>samtools)
./configure
make
make install
#注:编译安装默认路径为/usr/local/bin,所以无需添加环境变量
#测试安装
samtools 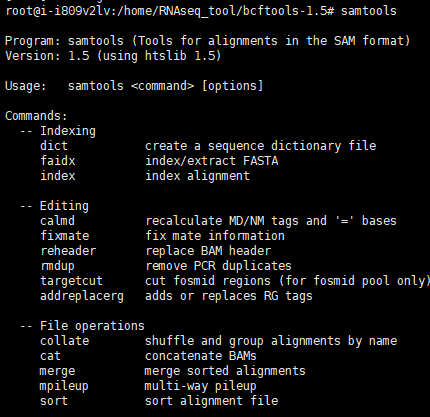
9.安装RSeQC(用于对bam文件进行质控,项目主页:http://rseqc.sourceforge.net/)
# Python2.7环境下
pip install RSeQC其由许多功能脚本组成,具体可以看官网信息(http://rseqc.sourceforge.net/)
10.安装HTseq(项目主页:http://htseq.readthedocs.io/en/release_0.9.1/)
# 安装依赖
sudo apt-get install build-essential python2.7-dev
python-numpy python-matplotlib python-pysam python-htseq
# Python2.7环境下
pip install HTSeq
#常用功能展示
htseq-count -h
11.安装bedtools
#下载并解压
wget https://github.com/arq5x/bedtools2/releases/download/v2.26.0/bedtools-2.26.0.tar.gz
tar –xzvf bedtools-2.26.0.tar.gz
#进入解压后文件目录进行编译安装
make
make install
#测试安装
bedtools --version![]()
12.安装cufflinks(旧流程)
#下载并解压
wget http://cole-trapnell-lab.github.io/cufflinks/assets/downloads/cufflinks-2.2.1.Linux_x86_64.tar.gz
tar –xzvf cufflinks-2.2.1.Linux_x86_64.tar.gz
#将目录添加至环境变量中
export PATH=$PATH:/home/RNAseq_tool/cufflinks-2.2.1.Linux_x86_64
#测试安装
cufflinks
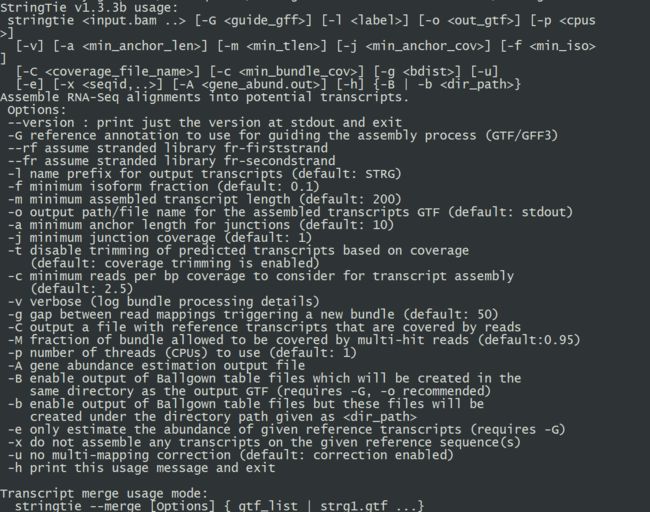
13.安装stringtie(cufflinks替代文件,项目主页:https://ccb.jhu.edu/software/stringtie/
#下载并解压
wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-1.3.3b.Linux_x86_64.tar.gz
tar -zxvf stringtie-1.3.3b.Linux_x86_64.tar.gz
#添加目录至系统环境变量
vim /etc/profile
添加如下行
export PATH=$PATH:/home/RNAseq_tool/stringtie-1.3.3b.Linux_x86_64
#试运行,如果如下图