EggNOG功能注释数据库在线和本地使用
文章目录
- COG简介
- eggNOG简介
- eggNOG mapper在线版
- eggNOG mapper本地版
- 安装说明
- 软件安装
- 数据库下载
- 基本使用
- HMMER方法
- diamond方法
- 结果解读
- 高级使用
- 服务器共用内存模式
- 宏基因组大数据模式
- 同源检索
- 功能注释
- 附1. emapper.py参数详解
- Reference
- 猜你喜欢
- 写在后面
COG简介
COG(Clusters of Orthologous Groups of proteins,直系同源蛋白簇)构成每个COG的蛋白都是被假定为来自于一个祖先蛋白,因此是orthologs或者是paralogs。
通过把所有完整基因组的编码蛋白一个一个的互相比较确定的。在考虑来自一个给定基因组的蛋白时,这种比较将给出每个其他基因组的一个最相似的蛋白(因此需要用完整的基因组来定义COG),这些基因的每一个都轮番的被考虑。如果在这些蛋白(或子集)之间一个相互的最佳匹配关系被发现,那么那些相互的最佳匹配将形成一个COG。这样,一个COG中的成员将与这个COG中的其他成员比起被比较的基因组中的其他蛋白更相像。
主页:https://www.ncbi.nlm.nih.gov/COG/

COG单字母描述,详见 http://www.sbg.bio.ic.ac.uk/~phunkee/html/old/COG_classes.html
COG one letter code descriptions
INFORMATION STORAGE AND PROCESSING
- [J] Translation, ribosomal structure and biogenesis
- [A] RNA processing and modification
- [K] Transcription
- [L] Replication, recombination and repair
- [B] Chromatin structure and dynamics
CELLULAR PROCESSES AND SIGNALING
- [D] Cell cycle control, cell division, chromosome partitioning
- [Y] Nuclear structure
- [V] Defense mechanisms
- [T] Signal transduction mechanisms
- [M] Cell wall/membrane/envelope biogenesis
- [N] Cell motility
- [Z] Cytoskeleton
- [W] Extracellular structures
- [U] Intracellular trafficking, secretion, and vesicular transport
- [O] Posttranslational modification, protein turnover, chaperones
METABOLISM
- [C] Energy production and conversion
- [G] Carbohydrate transport and metabolism
- [E] Amino acid transport and metabolism
- [F] Nucleotide transport and metabolism
- [H] Coenzyme transport and metabolism
- [I] Lipid transport and metabolism
- [P] Inorganic ion transport and metabolism
- [Q] Secondary metabolites biosynthesis, transport and catabolism
POORLY CHARACTERIZED
- [R] General function prediction only
- [S] Function unknown
eggNOG简介
eggNOG注释的原理和解读
通过已知蛋白对未知序列进行功能注释;
通过查看指定的eggNOG编号对应的protein数目,存在及缺失,从而能推导特定的代谢途径是否存在;
每个eggNOG编号是一类蛋白,将query序列和比对上的eggNOG编号的proteins进行多序列比对,能确定保守位点,分析其进化关系。
eggNOG mapper在线版
eggNOG-mapper就比对、注释eggNOG数据库的专用工具。
eggNOG-mapper在线分析,只需鼠标单击三步完成。
1.访问在线工具
http://eggnogdb.embl.de/#/app/emapper
2.参数设置
主要是选择蛋白序列文件,和设置邮箱。一般其它默认即可。

注意方法选择:diamond在序列少时相对较慢,但序列多时相对较快。HMMER方法对于亲源较远序列预测成功率更高,但数据量大时计算时间长,在线限制一次最多5000条序列。
3.提交任务
点击Run按扭即提交任务。会出现如下窗口。
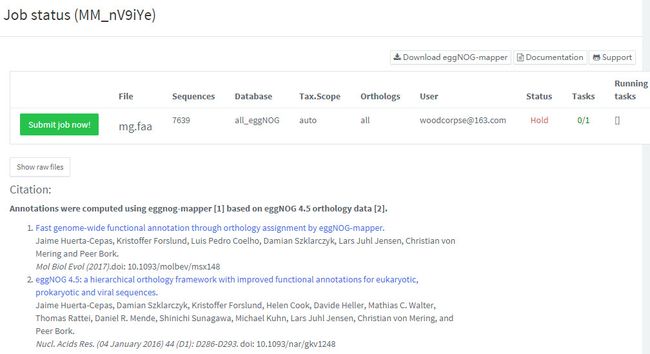
出现任务状态,和引文列表页面。值得注意的是,在线分析,即有序列限制,又要排队,如果用的人多,有时需要等很久。
eggNOG mapper本地版
更推荐conda安装,轻松稿定依赖关系和环境变量
conda install eggnog-mapper
手动软件下载和安装
cd ~/software
wget https://github.com/jhcepas/eggnog-mapper/archive/1.0.3.tar.gz
tar xvzf 1.0.3.tar.gz
cd eggnog-mapper-1.0.3
软件说明
less README.md
使用eggNOG数据库进行功能注释新基因、蛋白序列。常用于新基因组、转录组和宏基因组的基因集。直系同源(orthology)功能预测认为比传统的同源搜索更准确,可以避免直接从旁系同源(paralogs)借用功能注释(基因重复有很高的机会形成功能分化)。
帮助文档
https://github.com/jhcepas/eggnog-mapper/wiki
安装说明
软件依赖python2.7, wget, hmmer3, diamond,
硬盘空间要求:
- eggNOG注释数据库:~20GB
- eggNOG序列fasta文件:~20GB
- eggNOG数据库(euk, bact, arch): ~130GB,还有1-35GB的每个库对应的HMM数据库,不用全下载,需要什么下什么。
每个HMM库大小见 http://beta-eggnogdb.embl.de/download/eggnog_4.5/hmmdb_levels/
内存要求:
HMMER3注释时大内存时非常快,内存需要如下:
- 真核数据库euk: ~90GB
- 细菌数据库bact:~32GB
- 古细菌数据库arch:~10GB
软件安装
上面使用conda或wget下载方式安装,还可选git方式
git clone https://github.com/jhcepas/eggnog-mapper.git
数据库下载
- eggNOG提供了107个分类学的HMM数据库,三个最优数据库真核euk、细菌bact和古菌arch,和一个病毒特异数据库viruses
- 三个最优库包括对应所有HMM。
- 具体107个数据子集见 http://eggnogdb.embl.de/#/app/downloads
显示程序帮助
python eggnog-mapper/download_eggnog_data.py -h
下载四个常用数据库,保存于data目录。
指定程序下载至指定目录,并y自动同意,f强制下载
mkdir -p eggnog
python eggnog-mapper/download_eggnog_data.py --data_dir eggnog -y -f euk bact arch viruses
基本使用
cd eggnog-mapper
HMMER方法
本地检索细菌数据库
Disk based searches on the optimized bacterial database
-i输入、–output输出文件前缀、-d指定数据库数据、–data_dir指定数据库位置
python emapper.py -i test/polb.fa --output polb_bact -d bact --data_dir ~/data/db/eggnog
diamond方法
-m指定diamond方法,默认为hmmer方法。diamond在多于千条序列时才会体现速度优势,少量序列会感觉非常慢,而且结果也没有hmmer的更准确,尤其是对远源注释方面。
python emapper.py -i test/polb.fa --output diamond_bact_ -d bact --data_dir ~/data/db/eggnog -m diamond
时间较长,1个多小时
结果解读
https://github.com/jhcepas/eggnog-mapper/wiki/Results-Interpretation
结果有三个文件
polb_bact.emapper.annotations
polb_bact.emapper.hmm_hits
polb_bact.emapper.seed_orthologs
主要关注annotations结果,其中包括基因对应的GO、KEGG和COG描述
[project_name].emapper.hmm_hits文件:hmm比对结果列表
For each query sequence, a list of significant hits to eggNOG Orthologous Groups (OGs) is reported. Each line in the file represents a hit, where evalue, bit-score, query-coverage and the sequence coordinates of the match are reported. If multiple hits exist for a given query, results are sorted by e-value.
[project_name].emapper.seed_orthologs文件:最佳结果列表
each line in the file provides the best match of each query within the best Orthologous Group (OG) reported in the [project].hmm_hits file, obtained running PHMMER against all sequences within the best OG. The seed ortholog is used to fetch fine-grained orthology relationships from eggNOG. If using the diamond search mode, seed orthologs are directly obtained from the best matching sequences by running DIAMOND against the whole eggNOG protein space.
[project_name].emapper.annotations文件:比对结果整理,这才是重点。
This file provides final annotations of each query. Tab-delimited columns in the file are:
制表符分隔的13列文件,如下:
- 序列名query_name: query sequence name
- eggNOG编号seed_eggNOG_ortholog: best protein match in eggNOG
- seed_ortholog_evalue: best protein match (e-value)
- seed_ortholog_score: best protein match (bit-score)
- 预测基因名predicted_gene_name: Predicted gene name for query sequences
- 逗号分隔的GO注释GO_terms: Comma delimited list of predicted Gene Ontology terms
- KO编号注释KEGG_KO: Comma delimited list of predicted KEGG KOs
- 代谢反应BiGG_Reactions: Comma delimited list of predicted BiGG metabolic reactions
- 注释物种范围Annotation_tax_scope: The taxonomic scope used to annotate this query sequence
- OG编号Matching_OGs: Comma delimited list of matching eggNOG Orthologous Groups
- best_OG|evalue|score: Best matching Orthologous Groups (only in HMM mode)
- COG分类COG functional categories: COG functional category inferred from best matching OG
- 模型注释eggNOG_HMM_model_annotation: eggNOG functional description inferred from best matching OG
高级使用
https://github.com/jhcepas/eggnog-mapper/wiki/Advanced-usage-and-tips
大内存和多线程加速
–usemem可读入全部数据进内存,可使用内存预测加载数据,–cpu可设置多线程,–override是强制覆盖结果,否则有结果文件会中止
python emapper.py -i test/polb.fa --output polb_bact --database bact --data_dir ~/data/db/eggnog --usemem --cpu 10 --override
# Total time: 11.8659 secs
服务器共用内存模式
先读入细菌库,数据库选择,仅能指定某一类数据库
python emapper.py --database bact --data_dir ~/data/db/eggnog --cpu 10 --servermode
需要时间读入数据
Waiting for server to become ready... localhost 51500
直到显示:
Server ready listening at localhost:51500 and using 10 CPU cores
Use `emapper.py -d bact:localhost:51500 (...)` to search against this server
再启动分析命令
–usemem可读入全部数据进内存,可使用内存预测加载数据,–cpu可设置多线程
python emapper.py -i test/polb.fa --output polb_bact --database bact:localhost:51500 --data_dir ~/data/db/eggnog --usemem --cpu 10 --override
# Total time: 9.77332 secs
宏基因组大数据模式
https://github.com/jhcepas/eggnog-mapper/wiki/Setting-up-large-scale-analyses
大基因组,和宏基因组数据的注释(>100M的蛋白)。
分析主要分两步:同源检索,计算密集;功能注释,读写密集。数据拆分会提高效率。
同源检索
1. 序列拆分
准备文件并调整为单行fasta
cp /mnt/bai/yongxin/test/meta1809/temp/23prokka_all/mg.faa input_file0.faa
format_fasta_1line.pl -i input_file0.faa -o input_file.faa
拆分为文件,每个2百万行,1百万条序列。这里测序用10000行,5000条序列。
# -l按行数分割,-a后缀宽度3位,默认2位;-d数据后缀
split -l 10000 -a 3 -d input_file.faa input_file.chunk_
2.并行比对
方法1. 产生命令用于集群
for f in *.chunk_*; do
echo ./emapper.py -m diamond --no_annot --no_file_comments --cpu 16 -i $f -o $f;
done
方法2. 并行计算
time parallel -j 3 --xapply \
'python emapper.py -m diamond --no_annot --no_file_comments --data_dir ~/data/db/eggnog --cpu 16 -i {1} -o {1}' \
::: input_file.chunk*
耗时 real 14m45.579s
功能注释
此步为硬盘密集型,推荐将eggnog.db存储于SSD硬盘,或/dev/shm内存目录中
3. 合并比对结果
cat *.chunk_*.emapper.seed_orthologs > input_file.emapper.seed_orthologs
4.注释
为了提高速度,将数据库复制到内存,21s
cp ~/data/db/eggnog/eggnog.db /dev/shm
time emapper.py --annotate_hits_table input_file.emapper.seed_orthologs --no_file_comments -o output_file --cpu 20 --data_dir /dev/shm --override
数据库在内存时,处理1万条序列大约15s
现在我们获得了所有基因注释的列表。配合基因丰度矩阵,可以进行可种汇总、差异比较、功能描述了。
附1. emapper.py参数详解
python emapper.py -h
usage: emapper.py [-h] [--guessdb] [--database] [--dbtype {hmmdb,seqdb}]
[--data_dir] [--qtype {hmm,seq}] [--tax_scope]
[--target_orthologs {one2one,many2one,one2many,many2many,all}]
[--excluded_taxa]
[--go_evidence {experimental,non-electronic}]
[--hmm_maxhits] [--hmm_evalue] [--hmm_score]
[--hmm_maxseqlen] [--hmm_qcov] [--Z] [--dmnd_db DMND_DB]
[--matrix {BLOSUM62,BLOSUM90,BLOSUM80,BLOSUM50,BLOSUM45,PAM250,PAM70,PAM30}]
[--gapopen GAPOPEN] [--gapextend GAPEXTEND]
[--seed_ortholog_evalue] [--seed_ortholog_score] [--output]
[--resume] [--override] [--no_refine] [--no_annot]
[--no_search] [--report_orthologs] [--scratch_dir]
[--output_dir] [--temp_dir] [--no_file_comments]
[--keep_mapping_files] [-m {hmmer,diamond}] [-i]
[--translate] [--servermode] [--usemem] [--cpu]
[--annotate_hits_table] [--version]
optional arguments:
-h, --help 显示帮助show this help message and exit
--version 版本号
Target HMM Database Options:
--guessdb 根据物种ID猜所属数据库guess eggnog db based on the provided taxid
--database , -d 数据库选择,仅能指定某一类数据库specify the target database for sequence searches.Choose among: euk,bact,arch, host:port, or a local hmmpressed database
--dbtype {hmmdb,seqdb} 数据库类型
--data_dir 数据目录 Directory to use for DATA_PATH.
--qtype {hmm,seq} 方法选择,序列少用hmm,序列多用seq
Annotation Options:
--tax_scope 设定物种范围,默认自动调整Fix the taxonomic scope used for annotation, so only orthologs from a particular clade are used for functional transfer. By default, this is automatically adjusted for every query sequence.
--target_orthologs {one2one,many2one,one2many,many2many,all}
功能注释类型 defines what type of orthologs should be used for functional transfer
--excluded_taxa (for debugging and benchmark purposes)
--go_evidence {experimental,non-electronic}
注释准确度,只选实验 Defines what type of GO terms should be used for
annotation:experimental = Use only terms inferred from
experimental evidencenon-electronic = Use only non-
electronically curated terms
HMM search_options:
--hmm_maxhits 匹配结果数量,默认1 Max number of hits to report. Default=1
--hmm_evalue E-value threshold. Default=0.001
--hmm_score Bit score threshold. Default=20
--hmm_maxseqlen 忽略序列大于5000的蛋白Ignore query sequences larger than `maxseqlen`.
Default=5000
--hmm_qcov min query coverage (from 0 to 1). Default=(disabled)
--Z Fixed database size used in phmmer/hmmscan (allows
comparing e-values among databases).
Default=40,000,000
diamond search_options:
--dmnd_db DMND_DB 数据库位置Path to DIAMOND-compatible database
--matrix {BLOSUM62,BLOSUM90,BLOSUM80,BLOSUM50,BLOSUM45,PAM250,PAM70,PAM30}
Scoring matrix
--gapopen GAPOPEN Gap open penalty
--gapextend GAPEXTEND
Gap extend penalty
Seed ortholog search option:
--seed_ortholog_evalue
Min E-value expected when searching for seed eggNOG
ortholog. Applies to phmmer/diamond searches. Queries
not having a significant seed orthologs will not be
annotated. Default=0.001
--seed_ortholog_score
Min bit score expected when searching for seed eggNOG
ortholog. Applies to phmmer/diamond searches. Queries
not having a significant seed orthologs will not be
annotated. Default=60
Output options:
--output , -o base name for output files
--resume Resumes a previous execution skipping reported hits in
the output file.
--override Overwrites output files if they exist.
--no_refine Skip hit refinement, reporting only HMM hits.
--no_annot Skip functional annotation, reporting only hits
--no_search Skip HMM search mapping. Use existing hits file
--report_orthologs The list of orthologs used for functional transferred
are dumped into a separate file
--scratch_dir Write output files in a temporary scratch dir, move
them to final the final output dir when finished.
Speed up large computations using network file
systems.
--output_dir Where output files should be written
--temp_dir Where temporary files are created. Better if this is a
local disk.
--no_file_comments No header lines nor stats are included in the output
files
--keep_mapping_files Do not delete temporary mapping files used for
annotation (i.e. HMMER and DIAMOND search outputs)
Execution options:
-m {hmmer,diamond} 运行选项,默认为hmmer,可选diamondDefault:hmmer
-i 输入文件 Input FASTA file containing query sequences
--translate 输入核酸序列,翻译为蛋白 Assume sequences are genes instead of proteins
--servermode 数据载入内存模式,方便反复使用Loads target database in memory and keeps running in
server mode, so another instance of eggnog-mapper can
connect to this sever. Auto turns on the --usemem flag
--usemem 读入整个数据库至内存 If a local hmmpressed database is provided as target
using --db, this flag will allocate the whole database
in memory using hmmpgmd. Database will be unloaded
after execution.
--cpu 多线程
--annotate_hits_table
注释结果 Annotatate TSV formatted table of query->hits. 4
fields required: query, hit, evalue, score. Implies
--no_search and --no_refine.
Reference
https://github.com/jhcepas/eggnog-mapper/wiki
[1] Fast genome-wide functional annotation through orthology assignment by
eggNOG-mapper. Jaime Huerta-Cepas, Kristoffer Forslund, Luis Pedro Coelho,
Damian Szklarczyk, Lars Juhl Jensen, Christian von Mering and Peer Bork.
Mol Biol Evol (2017). doi:
10.1093/molbev/msx148
[2] eggNOG 4.5: a hierarchical orthology framework with improved functional
annotations for eukaryotic, prokaryotic and viral sequences. Jaime
Huerta-Cepas, Damian Szklarczyk, Kristoffer Forslund, Helen Cook, Davide
Heller, Mathias C. Walter, Thomas Rattei, Daniel R. Mende, Shinichi
Sunagawa, Michael Kuhn, Lars Juhl Jensen, Christian von Mering, and Peer
Bork. Nucl. Acids Res. (04 January 2016) 44 (D1): D286-D293. doi:
10.1093/nar/gkv1248
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2300+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”

点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA