洛谷P1140 相似基因
题目背景
大家都知道,基因可以看作一个碱基对序列。它包含了444种核苷酸,简记作A,C,G,TA,C,G,TA,C,G,T。生物学家正致力于寻找人类基因的功能,以利用于诊断疾病和发明药物。
在一个人类基因工作组的任务中,生物学家研究的是:两个基因的相似程度。因为这个研究对疾病的治疗有着非同寻常的作用。
题目描述
两个基因的相似度的计算方法如下:
对于两个已知基因,例如AGTGATGAGTGATGAGTGATG和GTTAGGTTAGGTTAG,将它们的碱基互相对应。当然,中间可以加入一些空碱基-,例如:

这样,两个基因之间的相似度就可以用碱基之间相似度的总和来描述,碱基之间的相似度如下表所示:

那么相似度就是:(−3)+5+5+(−2)+(−3)+5+(−3)+5=9(-3)+5+5+(-2)+(-3)+5+(-3)+5=9(−3)+5+5+(−2)+(−3)+5+(−3)+5=9。因为两个基因的对应方法不唯一,例如又有:
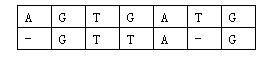
相似度为:(−3)+5+5+(−2)+5+(−1)+5=14(-3)+5+5+(-2)+5+(-1)+5=14(−3)+5+5+(−2)+5+(−1)+5=14。规定两个基因的相似度为所有对应方法中,相似度最大的那个。
输入输出格式
输入格式:
共两行。每行首先是一个整数,表示基因的长度;隔一个空格后是一个基因序列,序列中只含A,C,G,TA,C,G,TA,C,G,T四个字母。1≤1 \le 1≤序列的长度≤100 \le 100≤100。
输出格式:
仅一行,即输入基因的相似度。
输入输出样例
输入样例#1: 复制
7 AGTGATG
5 GTTAG
输出样例#1: 复制
14
#include
#include
using namespace std;
#define inf 0x3f3f3f3f
int value[5][5]={
{5,-1,-2,-1,-3},
{-1,5,-3,-2,-4},
{-2,-3,5,-2,-2},
{-1,-2,-2,5,-1},
{-3,-4,-2,-1,0}
};
string a,b;
int dp[102][102];
int tab(char c)
{
if(c=='A')return 0;
if(c=='C')return 1;
if(c=='G')return 2;
if(c=='T')return 3;
return 4;
}
int main()
{
int a1[102],b1[102];
int n,m;
cin>>n>>a;
cin>>m>>b;
for(int i=1;i<=n;i++)
a1[i]=tab(a[i-1]);
for(int i=1;i<=m;i++)
b1[i]=tab(b[i-1]);
for(int i=1; i<=n; i++)
for(int j=1; j<=m; j++)
dp[i][j]=-inf;
dp[0][0]=0;
for(int i=1; i<=n; i++) //初始化,这个为什么要初始化,具体还不太了解,看到别人的多了这一步ac了我也加上啦
dp[i][0]=dp[i-1][0]+value[a1[i]][4];//希望看到的大佬能告诉下我。
for(int j=1; j<=m; j++) //个人认为,毕竟i,j代表前i与前j匹配的最大值。
dp[0][j]=dp[0][j-1]+value[4][b1[j]];//故0的时候要初始化i,0的值和0,j的值,因为递推从这个基础上开始的。
//溜了,溜了,理解不深。献丑了
for(int i=1;i<=n;i++) //转移方程,三种情况递推。
{
for(int j=1;j<=m;j++)
{
dp[i][j]=max(dp[i-1][j-1]+value[a1[i]][b1[j]],max(dp[i][j-1]+value[4][b1[j]],dp[i-1][j]+value[a1[i]][4]));
}
}
cout << dp[n][m] << endl;
return 0;
}