MetaQuast:评估宏基因组拼接
文章目录
- MetaQuast:评估宏基因组拼接
- 热心肠日报
- 摘要
- 1 背景
- 2 材料和方法
- 2.1 基于参考的评估
- 2.2 从头评估
- 2.3 根据读长比对细化装配错误
- 2.3.1 SV检测
- 2.3.2 组装错误分类
- 2.4 可视化
- 图1. 基于MetaHIT数据集的HTML格式报告部分截图
- 结果
- Reference
- 猜你喜欢
- 写在后面

MetaQuast:评估宏基因组拼接
MetaQUAST: evaluation of metagenome assemblies
Bioinformatics, [4.531]
2015-11-26 Method
DOI: https://doi.org/10.1093/bioinformatics/btv697
第一作者:Alla Mikheenko
通讯作者:Alexey Gurevich
其它作者:Vladislav Saveliev
作者主要单位:
圣彼得堡国立大学转化生物医学研究所算法生物技术中心,圣彼得堡199034,俄罗斯(Center for Algorithmic Biotechnology, Institute of Translational Biomedicine, St. Petersburg State University, St. Petersburg 199034, Russia)
热心肠日报
- MetaQUAST是一款专门针对宏基因组拼接结果评估的工具;
- 评估主要步骤包括比对参考序列确定未知物种含量,提供基于多样性参考基因组的综合报告,通过检测嵌合重叠群确定是否存在高度相关的物种;
- 通过4种常用拼接工具分析一个模拟数据集和两个真实数据集的结果进行评估,测试结果表明MetaQUAST性能良好,同时发现没有一个软件在各方面都能优于其它软件;
- 该软件可为用户选择适合的拼接工具提供指导。
点评:宏基因组拼接软件众多,但由于缺少参考数据库,拼接结果评估困难。QUAST是2013年发表于Bioinformatics,是一款非常流行的基因组拼接结果评估软件,引用1759次。2016年又推出了专门针对宏基因组的MetaQUAST版本,引用125次(引用统计截止19年9月17日)。
摘要
简介:在过去的几年中,我们目睹了新的宏基因组拼接方法的快速发展。 尽管有许多针对单基因组装配的基准实用程序,但是没有公认的用于宏基因组特异性类似物的评估和比较工具。 在本文中,我们提出了MetaQUAST,它是QUAST的一种修改版本,是基于重叠群与参照对齐的基因组拼接评估的最先进工具。 MetaQUAST通过检测这些宏基因组数据集的特征:(i)未知物种含量通过与下载的参考序列比对来确定;(ii)提供巨大多样性的多个基因组的综合报告;(iii)通过检测嵌合重叠群而存在高度相关的物种。 我们通过比较一个模拟数据集和两个真实数据集上的几个主要组装软件来演示MetaQUAST性能。
可用性和实施:http://quast.sourceforge.net/metaquast
软件主页,已经更新至3.2版本,网站也更新,并随整合为QUAST中的一部分

对四种宏基因组拼接软件基于MetaHIT数据组装结果比较
联系方式:[email protected]
1 背景
1 Introduction
宏基因组学研究直接取自环境样品的遗传物质。 NGS技术允许甚至从低丰度生物体中测序短读长而无需克隆。然而,在这些实验中产生的数据往往是巨大的,嘈杂的,并且包含来自数千种物种的片段,其丰度和同源性变化很大。这些挑战导致了宏基因组装的新计算问题,其次是多种方法(Boisvert等,2012; Peng等,2012; Haider等,2014),这需要标准的基准程序进行比较。
大多数现有的组装评估方法不适用于宏基因组。然而,存在计算关于组装的读长可能性的方法(Clark等人,2013; Ghodsi等人,2013),或确定单拷贝保守的普遍存在的基因含量(Parks等人,2015; Simao等人,J.Biol.Chem.2007,1987)。 ,2015)。不幸的是,没有一个使用重叠群比对与密切相关的参考基因组。在本文中,我们介绍了MetaQUAST,这是一种基于QUAST的宏基因组特异性修改版软件(Gurevich等,2013)。 QUAST基于与给定的密切相关的参考基因组的比对来检测错误,并且还报告和绘制诸如N50和基因含量的重叠群统计数据,其甚至在没有用户提供参考序列的情况下给出了组成物种的概述。为了解释宏基因组拼接,MetaQUAST增加了几个新功能:(i)使用无限数量参考基因组的能力,(ii)自动物种内容检测,(iii)嵌合重叠群的检测(种间错误组装)和(iv)显著的重新设计
2 材料和方法
2.1 基于参考的评估
有充分研究的具有已知物种含量的宏基因组数据集(Qin等,2010)或模拟数据(Boisvert等,2012; Namiki等,2012)。它们可与MetaQUAST一起用于评估基于参考基因组比对的装配方法。多参考数据库的流程包括以下四个主要步骤(附图S1):
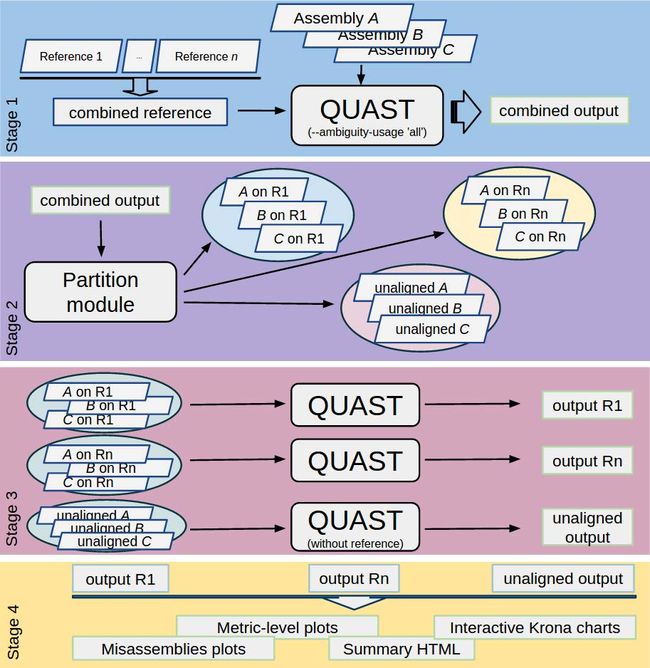
附图1. 基于参考数据库的评估流程
MetaQUAST pipeline for reference-based evaluation
-
所有参考基因组都连接成一个文件(组合参考)。 QUAST输入所有输入组装结果与参考数据。我们强制QUAST报告所有不确定的比对,而不是仅报告一个。对于包含密切相关物种的宏基因组数据集,所有模糊比对都是必不可少的。
-
我们将所有重叠群分成组,每组包含映射到特定参考基因组的序列(基于先前产生的比对)。映射到几个基因组的重叠群的组。无法比对的重叠群被放入一个额外的组中。
-
接下来,分别为每个输入参考数据库分别运行QUAST,为其提供相应的一组重叠群。无法比对的重叠群组不再进行比对。
-
最后,所有QUAST运行的结果将组合在一起,形成摘要报告和可视化。用户可以查看每次运行的详细完整QUAST输出,以及整个数据集结果的概览。
除了QUAST标准质量统计数据集(N50,基因组比例genome fraction等)之外,我们还添加了两个指标:
-
种间易位(interspecies translocations)数量:一种错误组装,其中侧翼序列与不同的参考序列对齐[类似于(Gurevich等人2013)中引入的易位,其中侧翼序列与不同的染色体对齐]。
-
可能错误组装的重叠群的数量:包含重叠群大部分对齐和未对齐片段的数量,因此可能包含具有未知基因组的种间易位。
与使用GeneMarkS的常规QUAST相比,MetaQUAST使用MetaGeneMark(Zhu等,2010)进行基因预测,该基因预测是专门为宏基因组开发的。
2.2 从头评估
2.2 De novo evaluation
多数实验宏基因组学研究使用的是从头组装,而没有参考信息。在没有输入参考序列或物种列表的情况下执行MetaQUAST时,它将尝试识别物种含量并自动提取参考序列。请注意,该算法在假设研究人员对微生物群落最感兴趣的前提下工作,因此搜索仅限于细菌和古细菌。
工作流程(请参见附图S2)首先应用BLASTn(Camacho等,2009)将重叠群与SILVA数据库中的16S rRNA序列进行比对(Quast等,2012)。几乎所有微生物物种中都存在的16S亚基是高度保守的序列,但还包括一个高变区,可用于将生物分类。对于每个检测到的物种,具有最高评分的一个菌株将保留在组装中。
查询针对NCBI的物种对对应丰度的前50名,下载每个物种的最少片段的序列。由于已知的问题与生物之间的rRNA操纵子的拷贝数不同以及16S基因的基因组内部异质性不同,某些下载的基因组序列可能在所评估的组装中不具有代表性。 MetaQUAST尝试通过除去重叠群覆盖率小于10%(对于所有组装)的基因组来过滤假阳性。在特殊情况下,当所有序列的基因组分数都非常低时,该列表将保持未经过滤的状态。
结果,我们获得了可能由组装序列代表的一组基因组。我们使用这些序列(如2.1节中所示)启动MetaQUAST,并产生与常规基于参考的分析相同的输出文件。
我们的方法是准确性和时间/内存消耗之间的折衷。为了获得更精确的结果,我们建议使用MGTAXA(Williamson et al。,2012)或基于精确读长比对的方法,例如Kraken(Wood and Salzberg,2014)或CLARK(Ounit et al。,2015)。通过对整个NCBI-nr数据库进行BLASTx(Altschul等,1990)搜索可以获得非常精确的结果。所获取的物种名称列表可以以纯文本格式输入到MetaQUAST,使其从NCBI数据库下载指定的序列,并将其用于基于参考的评估(请参阅第2.1节)。
2.3 根据读长比对细化装配错误
Refining misassemblies based on read mapping
常规的单基因组QUAST算法将重叠群和参考基因组之间的结构差异报告为错配。但是,在某些情况下,它们证明可能是结构变异(SV),而不是真实的装配错误。在分析没有相近参考序列的宏基因组学群落时,这一点尤其重要。 MetaQUAST通过考虑配对读长映射解决了这个问题(附图S3)。 MetaQUAST应用结构变异查找算法来基于不一致的读对检测断点,然后将其用于消除共享的断点breakpoints。
2.3.1 SV检测
SV detection
MetaQUAST利用bowtie2(Langmead等人,2009)对组合的参考基因组进行读长比对。 bowtie2生成的BAM文件(Li等,2009)按坐标排序,并作为SV发现软件的输入。我们选择了Manta(Chen等人,2015)SV挖掘软件,在我们的测试数据集上,其灵敏度和精度均优于LUMPY(Layer等人,2014)和Pindel(Ye等人,2009)。
2.3.2 组装错误分类
Misassembly classification
将QUAST报告的每个组装错误与所有发现的SV的断点置信区间进行比较。如果错误组装的开始和结束坐标都在SV间隔内扩展了一个小δ,则MetaQUAST会将此错误组装标记为假的,并且将不包括在最终报告中。如果在SV和错误组装之间未发现相似之处,则认为是真实的。默认δ值为100 bp,这是基于对真实和模拟数据集上出现的数十个SV进行手动分析的经验结果。
这种方法使我们能够显着减少所有三个测试数据集上错误报告的组装错误的数量。有关详细的基准测试结果,请参见补充材料。
2.4 可视化
Visualization
MetaQUAST通过大量鸟瞰图补充了QUAST可视化效果。此外,还将生成一个交互式摘要HTML报告,该报告结合了所有程序集和引用的关键统计信息。图表和摘要HTML在补充材料中进行了演示。
我们将汇总图分为三组:
-
错配
Misassembly图:按类型(错位relocations,倒位inversions,易位translocations和种间易位)进行错位分布。它们以两种视图形式存在:所有组装/参考序列和所有参考/组装。 -
公制级别
Metric-level的图:对于所有组装和所有参考序列,每个公制都有一个。基因组是从所有装配中的平均值排序,从最佳开始。 -
Krona图表(Ondov等人,2011年):每个组装一张,整个数据集一张。圆图显示了分类概况。仅在从头评估模式下可用。
交互式摘要HTML报告汇总了所有统计信息,参考序列和组装的表和图。每个表格行均显示组合参考的值,并且可以展开以显示每个参考的值(请参见图1)。蓝色/红色热图强调离群值。
图1. 基于MetaHIT数据集的HTML格式报告部分截图
Part of a summary HTML report for the MetaHIT dataset.
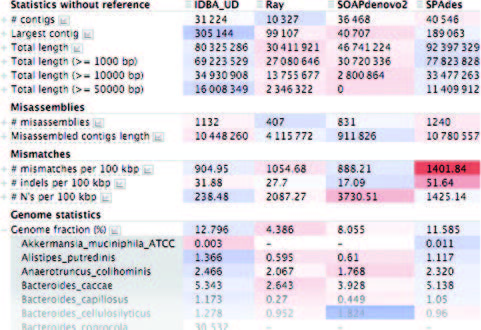
每个单元格按内容着色。在示例中,每个参考基因组比例的信息扩展开来。
结果
Results
我们在三个数据集上测试了MetaQUAST:CAMI(http://cami-challenge.org)模拟群落数据集,MetaHit的MH0045样本和HMP的SRS077736舌背女性样本(人类微生物组计划等,2012)。 我们使用在宏基因组学研究中常用的四种主要组装程序对这些数据进行拼接:IDBA-UD(Peng等人,2012),SPAdes(Bankevich等人,2012),Ray Meta(Boisvert等人,2012)和SOAPdenovo2(Luo等人,2012年)。 补充材料中展示了所有三个数据集的比较结果和MetaQUAST性能。
对这些数据集的比较表明,没有任何组装程序可以称为宏基因组学拼接中无可争议的领导者。 因此,诸如MetaQUAST之类的工具对群落而言具有重要的现实意义。 这将帮助科学家评估不同的组装软件,并为他们的研究选择最佳的分析。
Reference
Alexey Gurevich, Vladislav Saveliev, Nikolay Vyahhi & Glenn Tesler. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013, 29: 1072-1075. doi:10.1093/bioinformatics/btt086
Alla Mikheenko, Vladislav Saveliev & Alexey Gurevich. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2016, 32: 1088-1090. doi:10.1093/bioinformatics/btv697
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA