QIIME 2教程. 18序列双端合并read-joining(2020.2)
文章目录
- 前情提要
- 序列双端合并的另一种方法`read-joining`
- 数据下载
- 序列合并
- 查看合并序列的数据质量和摘要
- 序列质控
- Deblur
- 查看Deblur特征表
- 导入双端合并的序列
- 导入序列
- 查看导入含质量读长数据的摘要
- Reference
- 译者简介
- 猜你喜欢
- 写在后面
前情提要
以下是前面几节的微信推送文章:
- NBT:QIIME 2可重复、交互式的微生物组分析平台
- 1简介和安装Introduction&Install
- 2插件工作流程概述Workflow
- 3老司机上路指南Experienced
- 4人体各部位微生物组分析Moving Pictures,Genome Biology:人体各部位微生物组时间序列分析
- 5粪菌移植分析练习FMT,Microbiome:粪菌移植改善自闭症
- 6沙漠土壤分析Atacama soil,mSystems:干旱对土壤微生物组的影响
- 7帕金森小鼠教程Parkinson’s Mouse,Cell:肠道菌群促进帕金森发生ParkinsonDisease
- 8差异丰度分析gneiss
- 9数据导入Importing data
- 10数据导出Exporting data
- 11元数据Metadata
- 12数据筛选Filtering data
- 13训练特征分类器Training feature classifiers
- 14数据评估和质控Evaluating and controlling
- 15样品分类和回归q2-sample-classifier
- 16纵向和成对样本比较q2-longitudinal
- 17鉴定和过滤嵌合体序列q2-vsearch
序列双端合并的另一种方法read-joining
Alternative methods of read-joining in QIIME 2
https://docs.qiime2.org/2020.2/tutorials/read-joining/
注:最好按本教程顺序学习,想直接学习本章,至少完成本系列《1简介和安装》。
注意:本教程不包括DADA2的序列合并和去噪。相反,本教程重点介绍分析qiime 2中双端序列合并的替代方法。如果你有对DADA2去噪感和双端序列合并兴趣,《6沙漠土壤分析Atacama soil》教程演示了如何使用
qiime dada2 denoise-paired去噪双端序列。如果您计划使用DADA2来合并和消除双端数据的噪声,请在用DADA2去噪之前不要合并您的序列;DADA2希望读长尚未合并的序列,并将在去噪过程中为您双端合并。
在QIIME 2中,我们使用术语“单端序列”(single-end reads)单独指正向或反向序列;我们使用术语“双端序列”(paired-end reads)单独指尚未合并的正向和反向序列;并且我们使用术语“合并的序列(joined reads)”指已经联接(或合并)的正向和反向序列。理解这些术语中的哪一个适用于您的数据是很重要的,因为这将决定分析成对的最终数据需要哪些步骤。
目前,可以使用QIIME 2中的q2-vsearch插件合并双端序列,或者导入已在qiime 2之外合并的的序列(例如,使用fastq-join,有关详细信息,请参阅导入预合并的序列 Importing pre-joined reads)。本教程将涵盖这两个过程。
数据下载
Obtain the data
mkdir qiime2-read-joining-tutorial
cd qiime2-read-joining-tutorial
wget -c \
-O "demux.qza" \
"https://data.qiime2.org/2020.2/tutorials/read-joining/atacama-seqs.qza"
序列合并
Joining reads
# 11s
time qiime vsearch join-pairs \
--i-demultiplexed-seqs demux.qza \
--o-joined-sequences demux-joined.qza
输出对象:
demux.qza: 拆分后样本数据。 查看 | 下载demux-joined.qza:合并结果。 查看 | 下载
查看合并序列的数据质量和摘要
Viewing a summary of joined data with read quality
接下来我们获得拼接数据的可视化结果
qiime demux summarize \
--i-data demux-joined.qza \
--o-visualization demux-joined.qzv
输出可视化对象:
demux-joined.qzv: 可视化统计结果。 查看 | 下载
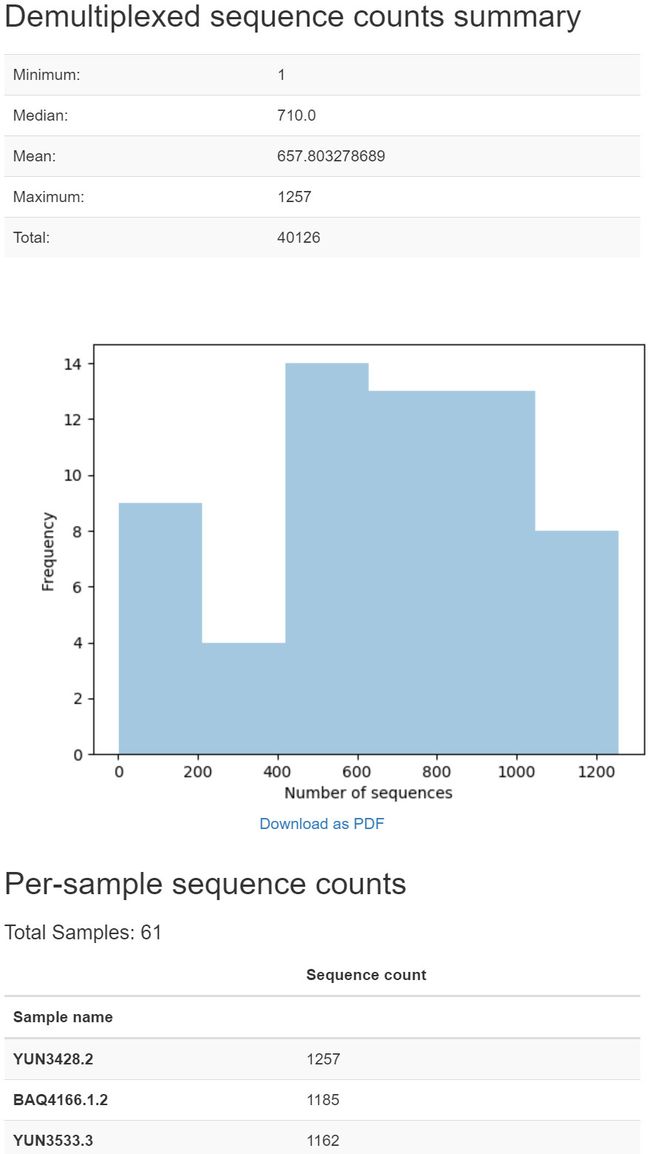

这份摘要报告对于确定你成功合并序列大约有多长特别有用(当我们用deblur去噪时,我们会回到这个问题上)。在这个可视化中查看质量图时,如果您将鼠标悬停在一个特定的位置上,将看到有多少个序列至少有那么长(为计算序列质量而采样的序列数量统计)。记下最高的序列位置,其中大部分(比如,>99%)的序列至少有那么长。
例如,当将鼠标悬停在可视化箱线图中的一个黑箱体上时(该黑箱体是由比本教程中使用的数据集更大的数据集生成的),我看到40126个序列中有10000个用于估计该位置的质量分数分布。
当我将鼠标悬停在位置250(用红色方框表示)上时,我看到一些序列没有这么长,因为只有9994个序列用于估计该位置的质量分数分布。下面的红色框和红色文本告诉我,有些序列没有这么长。
当我将鼠标悬停在254号位置(也用一个红框表示)上时,我看到许多序列没有这么长,因为只有845个序列用于估计该位置的质量分数分布。
基于对这些图的比较,我将注意到我的大多数序列至少有250个碱基长。我们计划在不久的将来简化这个过程。
序列质控
Sequence quality control
接下来,我们将使用质量过滤器quality-filter q-score-joined对序列进行质量控制。此方法与质量过滤 quality-filter q-score 相同,只是它仅对合并的序列进行操作。此方法的参数尚未在双端合并的数据上进行广泛的基准测试,因此我们建议尝试使用不同的参数设置。
# 18s
time qiime quality-filter q-score-joined \
--i-demux demux-joined.qza \
--o-filtered-sequences demux-joined-filtered.qza \
--o-filter-stats demux-joined-filter-stats.qza
输出对象:
demux-joined-filter-stats.qza: 统计结果。 查看 | 下载demux-joined-filtered.qza: 数据过滤后结果。 查看 | 下载
在这个阶段,您可以选择继续使用[Deblur](http://msystems.asm.org/content/2/2/e00191-16)进行额外的质量控制,或者您也可以进行序列去冗余,并选择使用q2-vsearch将它们聚类到OTU中。Deblur可以给出更高质量的结果,因此我们推荐该过程,并将在本教程的下一个步骤中说明该方法。
如果您有兴趣尝试一个更像QIIME 1处理的分析工作流(例如,要将Deblur或Dada2结果与QIIME 1类似的流程进行比较),那么接下来应该去冗余并聚类您的序列。如果您尝试此选项,我们强烈建议使用 qiime quality-filter q-score-joined 具有更高的最小质量阈值(--p-min-quality 20 或 --p-min-quality 30)(参见Bokulich等人2013年的文章学习更多细节)。然后,您可以按照OTU聚类教程中的步骤进行操作。在聚类之后,您可能希望使用qiime feature-table filter-features --p-min-samples筛选在至少一些样品中出现的特征。此外,还建议使用丰度过滤器去除单体(见Bokulich等人2013年的文章),以及过滤嵌合序列。
Deblur
你现在已经准备好用Deblur去噪你的序列了。您应该从质量分数图中为--p-trim-length选择合适的序列长度值。这将把所有序列修剪到这个长度,并丢弃任何小于这个长度的序列。
注释:我们使用的修剪长度为250,基于从教程数据集生成的质量分数图。不要将250与自己的数据集一起使用,因为该值将取决于数据集的序列长度。使用质量分数图为数据选择适当的修剪长度。
# 5m
time qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-joined-filtered.qza \
--p-trim-length 250 \
--p-sample-stats \
--o-representative-sequences rep-seqs.qza \
--o-table table.qza \
--o-stats deblur-stats.qza
输出对象:
rep-seqs.qza: 代表序列。 查看 | 下载deblur-stats.qza: 统计过程。 查看 | 下载table.qza: 特征表。 查看 | 下载
查看Deblur特征表
View summary of Deblur feature table
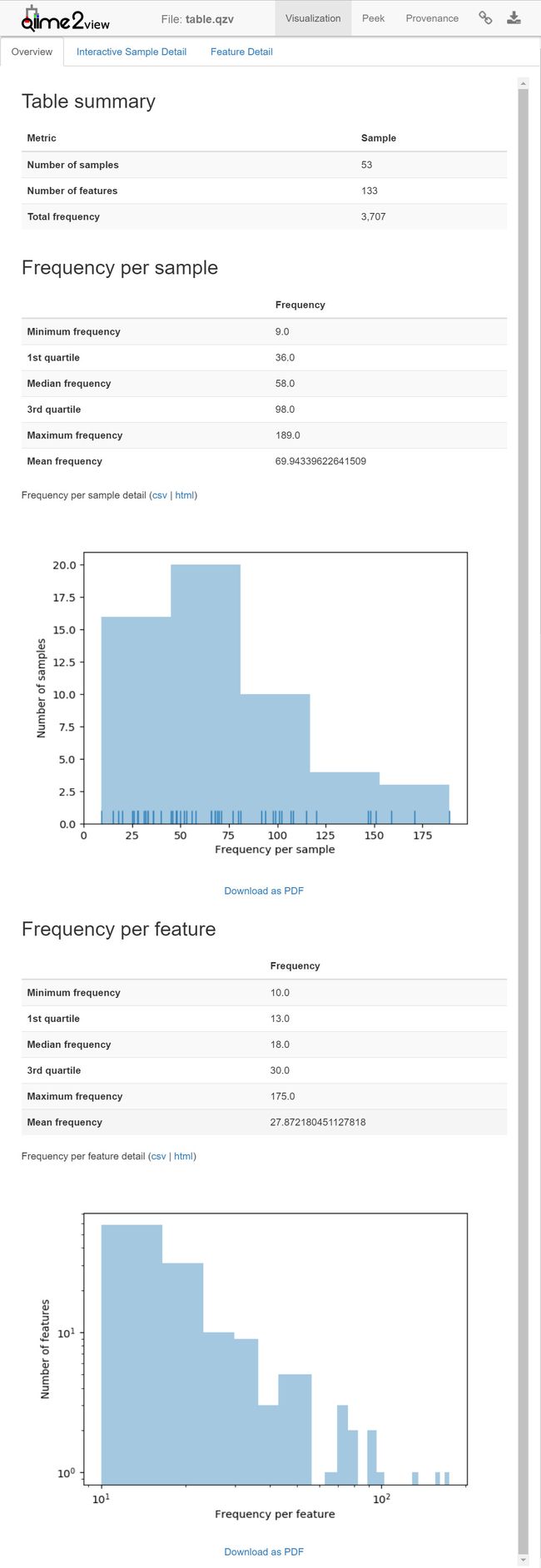
接下来,您可以总结q2-deblur生成的功能表。这个表和相应的代表序列现在可以用同样的方法和可视化工具来分析,这些方法和可视化工具将用于单端序列数据。
# 9s
time qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv
输出可视化对象:
table.qzv: 特征表可视化。 查看 | 下载
导入双端合并的序列
Importing pre-joined reads
下载测试数据并解压
wget -c \
-O "fj-joined.zip" \
"https://data.qiime2.org/2020.2/tutorials/read-joining/fj-joined.zip"
unzip fj-joined.zip
导入序列
Import reads
使用 qiime tools import 导入数据,使用的数据格式为 SingleEndFastqManifestPhred33 。在将来的升级中,我们将来升级的清晰描述为一种合并的序列数据。但是在当下,你应该采用单端Fastq Mainfest格式导入。
# 7s
time qiime tools import \
--input-path fj-joined/manifest \
--output-path fj-joined-demux.qza \
--type SampleData[JoinedSequencesWithQuality] \
--input-format SingleEndFastqManifestPhred33
输出结果:
fj-joined-demux.qza:导入的合并双端序列。 查看 | 下载
查看导入含质量读长数据的摘要
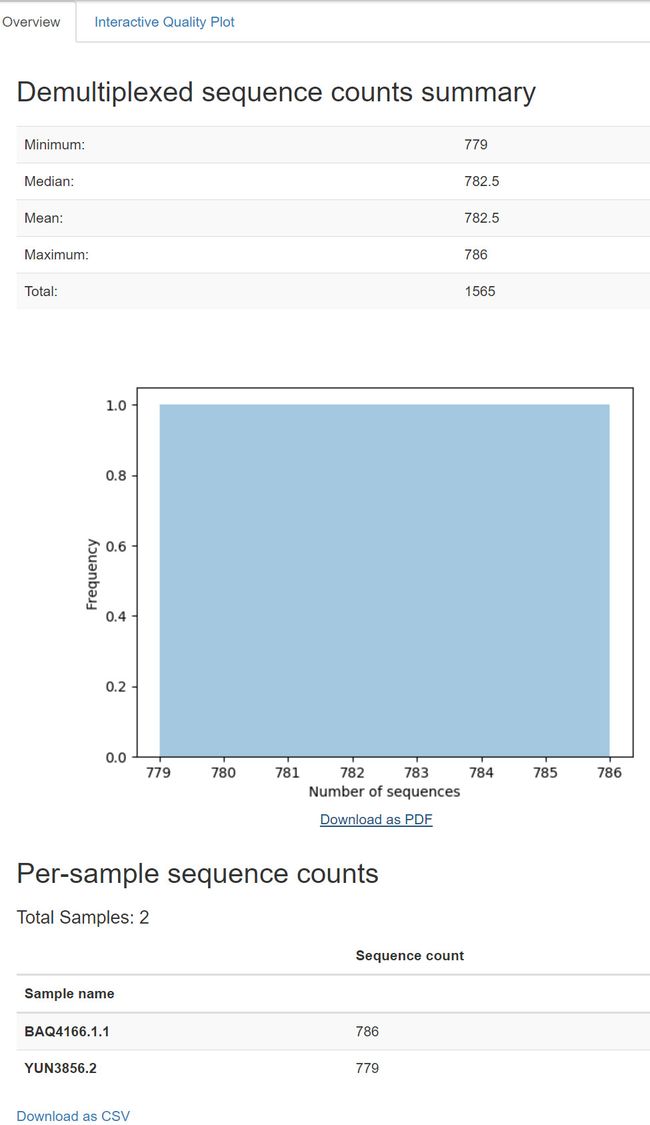
Viewing summary of imported data with read quality
qiime demux summarize \
--i-data fj-joined-demux.qza \
--o-visualization fj-joined-demux.qzv
输出结果:
fj-joined-demux.qzv:导入的合并双端序列的摘要。 查看 | 下载
现在你可以使用上面的方法继续分析此数据了,使用q2-quality-filter质控, q2-deblur去噪, 或q2-vsearch去冗余和挑选OTU。
祝你QIIME使用愉快!
Reference
https://docs.qiime2.org/2020.2/
Evan Bolyen*, Jai Ram Rideout*, Matthew R. Dillon*, Nicholas A. Bokulich*, Christian C. Abnet, Gabriel A. Al-Ghalith, Harriet Alexander, Eric J. Alm, Manimozhiyan Arumugam, Francesco Asnicar, Yang Bai, Jordan E. Bisanz, Kyle Bittinger, Asker Brejnrod, Colin J. Brislawn, C. Titus Brown, Benjamin J. Callahan, Andrés Mauricio Caraballo-Rodríguez, John Chase, Emily K. Cope, Ricardo Da Silva, Christian Diener, Pieter C. Dorrestein, Gavin M. Douglas, Daniel M. Durall, Claire Duvallet, Christian F. Edwardson, Madeleine Ernst, Mehrbod Estaki, Jennifer Fouquier, Julia M. Gauglitz, Sean M. Gibbons, Deanna L. Gibson, Antonio Gonzalez, Kestrel Gorlick, Jiarong Guo, Benjamin Hillmann, Susan Holmes, Hannes Holste, Curtis Huttenhower, Gavin A. Huttley, Stefan Janssen, Alan K. Jarmusch, Lingjing Jiang, Benjamin D. Kaehler, Kyo Bin Kang, Christopher R. Keefe, Paul Keim, Scott T. Kelley, Dan Knights, Irina Koester, Tomasz Kosciolek, Jorden Kreps, Morgan G. I. Langille, Joslynn Lee, Ruth Ley, Yong-Xin Liu, Erikka Loftfield, Catherine Lozupone, Massoud Maher, Clarisse Marotz, Bryan D. Martin, Daniel McDonald, Lauren J. McIver, Alexey V. Melnik, Jessica L. Metcalf, Sydney C. Morgan, Jamie T. Morton, Ahmad Turan Naimey, Jose A. Navas-Molina, Louis Felix Nothias, Stephanie B. Orchanian, Talima Pearson, Samuel L. Peoples, Daniel Petras, Mary Lai Preuss, Elmar Pruesse, Lasse Buur Rasmussen, Adam Rivers, Michael S. Robeson, Patrick Rosenthal, Nicola Segata, Michael Shaffer, Arron Shiffer, Rashmi Sinha, Se Jin Song, John R. Spear, Austin D. Swafford, Luke R. Thompson, Pedro J. Torres, Pauline Trinh, Anupriya Tripathi, Peter J. Turnbaugh, Sabah Ul-Hasan, Justin J. J. van der Hooft, Fernando Vargas, Yoshiki Vázquez-Baeza, Emily Vogtmann, Max von Hippel, William Walters, Yunhu Wan, Mingxun Wang, Jonathan Warren, Kyle C. Weber, Charles H. D. Williamson, Amy D. Willis, Zhenjiang Zech Xu, Jesse R. Zaneveld, Yilong Zhang, Qiyun Zhu, Rob Knight & J. Gregory Caporaso#. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology. 2019, 37: 852-857. doi:10.1038/s41587-019-0209-9
译者简介
刘永鑫,博士。2008年毕业于东北农大微生物学,2014年于中科院遗传发育所获生物信息学博士,2016年遗传学博士后出站留所工作,任宏基因组学实验室工程师。目前主要研究方向为微生物组数据分析、分析方法开发与优化和科学传播,QIIME 2项目参与人。目前在***Science、Nature Biotechnology、Cell Host & Microbe、Current Opinion in Microbiology*** 等杂志发表论文20余篇。2017年7月创办“宏基因组”公众号,目前分享宏基因组、扩增子原创文章500余篇,代表博文有《扩增子图表解读、分析流程和统计绘图三部曲(21篇)》、《Nature综述:手把手教你分析菌群数据(1.8万字)》、《QIIME2中文教程(22篇)》等,关注人数8万+,累计阅读1300万+。
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA