蛋白质配体复合物-分子动力学模拟Gromacs
前言
本人菜鸟,记录一下自己学习使用Gromacs的过程。
蛋白质配体复合物模拟一般过程:
获取蛋白质结构文件、分子对接后的配体结构文件 → 准备蛋白质拓扑 → 选择力场 → 小分子拓扑 → 复合物拓扑 → 添加盒子、溶剂 → 添加离子 → 能量最小化 → NVT、NPT平衡 → 开始模拟 → 分析结果
软件:gromacs2021
目录
1.准备蛋白质结构文件、分子对接后的配体结构文件
2.准备拓扑、选择力场
3.小分子拓扑
4.创建复合物拓扑
5.添加盒子、溶剂
6.添加离子
7.能量最小化
8.NVT、NPT平衡
9.开始模拟
10.分析结果
开始
1.准备蛋白质结构文件、分子对接后的配体结构文件
首先得到分子对接后的蛋白质结构文件与配体结构文件(通常得分最高的或你选择的构象)

2.准备蛋白质拓扑、选择力场
在服务器上输入如下命令
gmx pdb2gmx -f CBHI.pdb -o CBHI_processed.gro -water spcgmx会提示选择一个力场,输入6,选择Amber99sb-ildn力场
注:如果出现如下报错
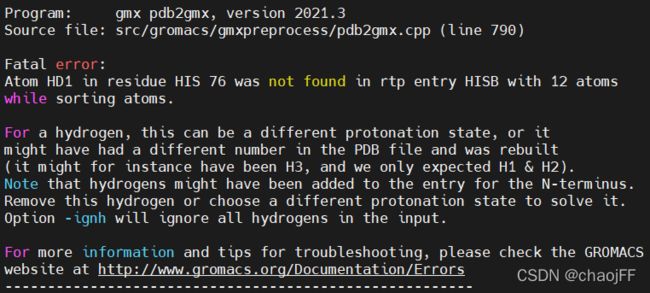
说明pdb文件中对氢原子的命名与力场rtp文件中命名规则与顺序不同。解决方法:①如果需要保留初始氢原子,需要找到该力场的rtp文件查看文件中的命名规则对氢原子重命名。②在上面的命令行中末尾加上 -ignh 命令,用此选项忽略文件中的氢原子, 统一使用氢原子命名规则, 以免出现氢原子名称不一致的情况。
选择完力场得到三个文件.gro, topol.top和.itp

3.手动小分子拓扑
由于力场无法自动处理配体,我们需要第三方的方法获得配体的力场参数,下面使用两种方法。
3a
使用ACPYPE在线版处理配体,进入网站Bio2Byte home page
提交之后
3b
使用ACPYPE离线版处理配体
使用AmberTools中的antechamber模块可以快速地生成所需要的配体分子原子电荷以及定义原子类型,通过以下命令可调用antechamber:
antechamber -i Ligand_Rank1.pdb -fi pdb -o PNPC.mol2 -fo mol2 -c bcc -nc 0 -rn PNP -at gaff2得到PNPC.mol2
注:
-at gaff2:指定使用GAFF2力场;
-c bcc:使用AM1-BCC2方法计算原子电荷;
-nc 0:指的是分子的净电荷为0;
-rn PNP:指定小分子的残基名为“PNP”;
-fo mol2:输出的mol2文件中保留原子类型以及原子电荷。
使用acpype.py处理配体
acpype.py -i PNPC.mol2 -n 0 -d运行完成后,在当前目录下出现一个“PNPC.acpype”的文件夹,文件夹中的“PNPC_GMX.itp”与“PNPC_GMX.gro”为gromacs所需要的文件。
![]()
4.创建复合物拓扑
4a
将之前处理得到的蛋白质文件(CBHI_processed.gro)复制并改名为complex.gro
cp CBHI_processed.gro complex.gro接下来复制PNPC_GMX.gro的坐标部分粘贴到complex.gro文件中蛋白质原子最后一行的下面
由于添加了57个原子到.gro文件中, 将complex.gro文件中第二行的数字增加57
4b
在topol.top文件中插入一行,内容为
; Include ligand topology
#include "PNPC_GMX.itp"
如图

最后在文档的末尾修改[ molecules ]部分,添加PNP信息

5.添加盒子、溶剂
定义单元盒子
gmx editconf -f complex.gro -o newbox.gro -bt dodecahedron -d 1.0得到newbox.gro 填充溶剂水
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro得到solv.gro文件
6.添加离子
点击下载em.mdp文件,存放到同工程目录下。
然后输入命令
gmx grompp -f em.mdp -c solv.gro -p topol.top -o next.tpr如果出现平衡电荷数警告,在命令后端加上 -maxwarn 1
gmx grompp -f em.mdp -c solv.gro -p topol.top -o next.tpr -maxwarn 1
翻看上面的输出信息,可以看到整个体系携带的电荷数。

由于生命体系中不存在净电荷,必须在体系中添加离子。
-pname 阳离子的名称 -nname 阴离子的名称 -np 阳离子个数 -nn 阴离子个数
gmx genion -s next.tpr -o solv_ions.gro -p topol.top -pname NA -np 19 运行上面的命令后会提示选择溶剂,选择15 SOL。
得到solv_ions.gro文件
5.能量最小化
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr得到em.tpr文件,运行EM。
gmx mdrun -v -deffnm em得到四个文件.edr .trr .log .gro
6.NVT、NPT平衡
点击下载nvt.mdp npt.mdp文件,存放到同工程目录下。
平衡蛋白质配体复合物与平衡任何其他蛋白质水溶液体系一样. 但需要有些特殊考虑:
a.对配体施加限制
b.处理温度耦合组
a.对配体施加限制
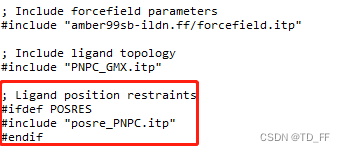
gmx genrestr -f PNPC_GMX.gro -o posre_PNPC.itp -fc 1000 1000 1000在topol.top文件中添加如下几行
; Ligand position restraints
#ifdef POSRES
#include "posre_PNPC.itp"
#endif
如图
b.热浴
对只有几个原子的组在控制其动能的涨落时, 温度耦合算法不够稳定,不要对体系中的每一单个物种使用独立的耦合。创建一个特殊的组, 其中包含蛋白质和配体。
gmx make_ndx -f em.gro -o index.ndx通过下面的命令合并"Protein"和"PNP",其中">"是make_ndx的提示符:
> 1 | 13
> q接下来开始nvt平衡
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr -n index.ndx运行之后得到nvt.tpr文件
gmx mdrun -deffnm nvtnext..
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr -n index.ndx运行之后得到nvt.tpr文件
gmx mdrun -deffnm npt7.开始模拟
依次运行命令
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr-v 可显示计算结束时间
gmx mdrun -deffnm md_0_1 -vGO!
注:
①如果要使用gpu加速,可在末尾加上 -nb gpu 来使用gpu加速计算,-v 可显示计算结束时间。
gmx mdrun -deffnm md_0_1 -nb gpu -v②如果模拟过程中因突发状况导致模拟终止,可使用如下命令续跑。
在同一个文件夹下运行(假设你的任务名称是 md_0_1)
gmx mdrun -s md_0_1.tpr -cpi md_0_1.cpt -deffnm md_0_18.分析结果
附官方使用翻译手册