分子动力学模拟Gromacs一般使用步骤(空蛋白)
前言
本人菜鸟,记录一下自己学习使用Gromacs的过程。
模拟一般过程:
获取蛋白质结构文件 → 准备拓扑 → 选择力场 → 添加盒子、溶剂 → 添加离子 → 能量最小化 → 温度、压力平衡 → 开始模拟 → 分析结果
软件:gromacs2021
目录
1.准备蛋白质结构文件
2.准备拓扑、选择力场
3.添加盒子、溶剂
4.添加离子
5.能量最小化
6.温度、压力平衡
7.开始模拟
8.分析结果
开始
1.准备蛋白质结构文件
蛋白质结构文件(.pdb)可以从PDB蛋白质数据库中下载(RCSB PDB: Homepage)
查找你需要的蛋白质

以1ETH为例
 下载PDB格式的文件
下载PDB格式的文件
注:蛋白质结构文件里会有水分子,可下载上图中PDB Format(gz)文件,将文件中的.ent文件用Discovery Studio或Pymol打开并去除水分子,并不是任何时候都需要进行这个过程(比如对与活性位点结合的水分子就不能去除)。
2.准备拓扑、选择力场
在服务器上输入如下命令
gmx pdb2gmx -f 1ETH.pdb -o 1ETH_processed.gro -water spcgmx会提示选择一个力场,输入8,选择CHARMM27力场
注:如果出现如下报错
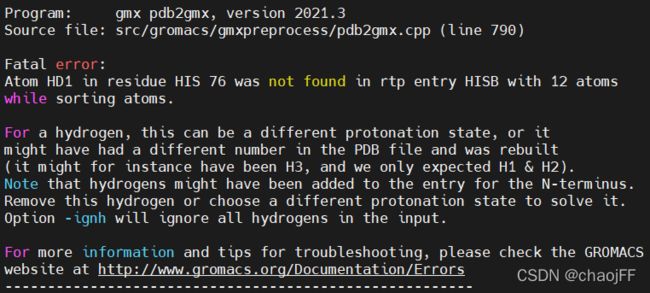
说明pdb文件中对氢原子的命名与力场rtp文件中命名规则与顺序不同。解决方法:①如果需要保留初始氢原子,需要找到该力场的rtp文件查看文件中的命名规则对氢原子重命名。②在上面的命令行中末尾加上 -ignh 命令忽略氢原子。
选择完力场得到三个文件.gro, topol.top和.itp

3.添加盒子、溶剂
定义单元盒子
gmx editconf -f 1ETH_processed.gro -o newbox.gro -bt dodecahedron -d 1.0得到newbox.gro 填充溶剂水
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solvate.gro得到solvate.gro文件
4.添加离子
点击下载em.mdp文件,存放到同工程目录下。
然后输入命令
gmx grompp -f em.mdp -c solvate.gro -p topol.top -o next.tpr如果出现平衡电荷数警告,在命令后端加上 -maxwarn 1
gmx grompp -f em.mdp -c solvate.gro -p topol.top -o next.tpr -maxwarn 1
翻看上面的输出信息,可以看到整个体系携带的电荷数。

由于生命体系中不存在净电荷,必须在体系中添加离子。
-pname 阳离子的名称 -nname 阴离子的名称 -np 阳离子个数 -nn 阴离子个数
gmx genion -s next.tpr -o solv_ions.gro -p topol.top -pname NA -np 8 -nname CL 运行上面的命令后会提示选择溶剂,选择13 SOL。
得到solv_ions.gro文件
5.能量最小化
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr得到em.tpr文件,运行EM。
gmx mdrun -v -deffnm em得到四个文件.edr .trr .log .gro
6.温度、压力平衡
点击下载nvt.mdp npt.mdp文件,存放到同工程目录下。
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr运行之后得到nvt.tpr文件
开始nvt
gmx mdrun -deffnm nvtnext..
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr运行之后得到npt.tpr文件
开始npt
gmx mdrun -deffnm npt7.开始模拟
点击下载md.mdp文件,存放到同工程目录下。
依次运行命令
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr-v 可显示计算结束时间
gmx mdrun -deffnm md_0_1 -v
GO!
注:
①如果要使用gpu加速,可在末尾加上 -nb gpu 来使用gpu加速计算,-v 可显示计算结束时间。
gmx mdrun -deffnm md_0_1 -nb gpu -v②如果模拟过程中因突发状况导致模拟终止,可使用如下命令续跑。
在同一个文件夹下运行(假设你的任务名称是 md_0_1)
gmx mdrun -s md_0_1.tpr -cpi md_0_1.cpt -deffnm md_0_18.分析结果
附官方使用手册翻译